














- DAZ.online
- DAZ / AZ
- DAZ 32/2022
- Wer entscheidet über den...

Foto: Dziurek/AdobeStock
Politik
Wer entscheidet über den Austausch?
Biologicals, Bioidenticals und Biosimilars – besser Switch als Substitution
Klar ist schon jetzt, dass sich sowohl Apotheker- als auch Ärzteverbände gegen das Prinzip des automatischen Austauschs, der sogenannten Substitution, wehren und eine ähnliche Problematik hinsichtlich Lieferfähigkeit und Adhärenz befürchten, wie wir es bereits im Zusammenhang mit den Rabattverträgen im Generikamarkt kennen.
Perspektive des Gesetzgebers
Während die politischen Entscheider in einer identischen Aminosäuresequenz offensichtlich auch einen identischen Wirkstoff sehen, sollten das die Fachkreise kritisch hinterfragen – was seit einiger Zeit auch getan wird. Biologika sind aufgrund ihres biologischen Herstellungsprozesses nicht so exakt und konkret definierbar wie die klassisch chemisch-synthetischen Wirkstoffe: lebende Organismen produzieren niemals exakte Kopien, stets kann eine inhärente Variabilität bestehen. Zudem besitzen Nebenprodukte, die beim Downstream-Prozess (siehe "Wie Biologika hergestellt werden") nicht vollständig abgetrennt werden, immer auch ein Potenzial für Immunogenität.
Doch welche Argumente werden für und wider einer Substitution, also des (automatischen) Austauschs von Biologika in der Apotheke genannt?
Beispiel aus der Apothekenpraxis
Im Rahmen der klinischen Entwicklung des Biosimilars Amgevita® (Adalimumab) wurden Daten in Phase-III-Studien an Patienten mit rheumatoider Arthritis und Psoriasis erhoben. Der pharmazeutische Unternehmer konnte jedoch im Rahmen der Zulassung das vollständige Indikationsfeld des Referenzarzneimittels Humira® für sich in Anspruch nehmen, welches neben rheumatoider Arthritis und Psoriasis auch Plaque-Psoriasis, Ankylosierende Spondylitis, Hidradenititis suppurativa, Morbus Crohn, Colitis ulcerosa und Uveitis umfasst.
Betrachtung aus regulatorischer Perspektive
Während die Entwicklung eines klassischen chemisch-synthetischen Generikums in der Regel nur eine Bioäquivalenzstudie erfordert, ist die Entwicklung eines Biosimilars wesentlich komplexer und erfordert ein sehr umfassendes und aufwendiges klinisches Entwicklungsprogramm.
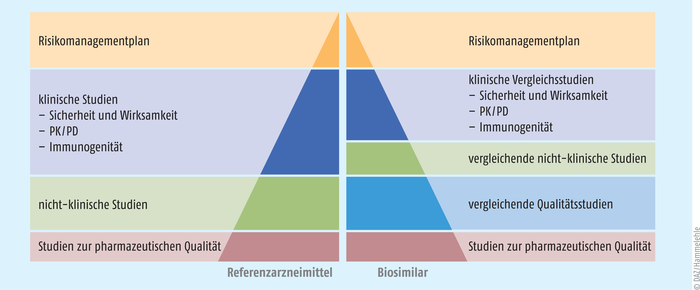
Vorteilhaft ist es, dass vergleichende (und somit weniger aufwendige) Studien zum Referenzarzneimittel zulässig und die Regel sind (s. Abb. 1). Im Rahmen des Zulassungsantrags sind den Behörden Studien bezüglich der Immunogenität, Pharmakokinetik und Pharmakodynamik (PK/PD) sowie zur Wirksamkeit und Sicherheit vorzulegen. Selbstverständlich werden auch die Daten zur pharmazeutischen Qualität und zu weiteren essenziellen Punkten gefordert. Als Zulassungsverfahren ist in der Regel nur das zentral europäische Verfahren der European Medicines Agency (EMA) möglich. Eine Ausnahme bilden hier einige niedermolekulare Heparine, die aus der Darmschleimhaut von Schweinen gewonnen werden, die noch im nationalen, dezentralen Verfahren zugelassen werden können (s. Tab. 1).
Eine Besonderheit bei der Zulassung von Biosimilars stellt das Verfahren der sogenannten Extrapolation dar. Hierbei sind mit dem Studienprogramm Wirksamkeit und Sicherheit für ein oder mehrere ausgewählte Indikationsgebiete wissenschaftlich zu belegen. Da die Referenzarzneimittel häufig über weitere Indikationsfelder verfügen, werden im Rahmen der Biosimilar-Zulassung auch diese nicht in den Studien erfassten Indikationsgebiete zur Anwendung des Fertigarzneimittels erteilt. Dies ist ein wesentlicher Unterschied zu den klassischen Generika, bei denen zur Erweiterung der Indikationsgebiete in der Regel Wirksamkeitsstudien an entsprechenden Patientenkohorten verlangt werden.
Abb. 1: Vergleich der Datenanforderungen für die europäische Zulassung von Biosimilars gegenüber dem jeweiligen Referenzarzneimittel (PK/PD: Pharmakokinetik und Pharmakodynamik) [Quelle: EMA]
Berechtigte Bedenken gegen den Austausch
Als biologische Produkte unterliegen die Wirkstoffe von Charge zu Charge, wie auch zwischen Referenzarzneimittel und Biosimilar, einer gewissen Variabilität bzw. Mikroheterogenität. Betroffen ist hier nicht die Primärstruktur, sondern allenfalls die räumliche Struktur (Tertiärstruktur) und vor allem das Glykosylierungsmuster. Bezüglich Wirksamkeit und Sicherheit sind seit 2006 und der ersten Anwendung von Biosimilars keine nennenswerten Probleme dokumentiert worden.
Die wesentliche Gefahr geht aber vom Faktor Mensch aus: Problematisch wird es vor allem dann, wenn sich die Applikationsmethode zwischen Referenzarzneimittel und Biosimilar unterscheidet. Doch auch abgesehen von solchen offensichtlichen Unterschieden bei der Präsentation und Handhabung eines Biosimilars gibt es weitere Stolperfallen. Ein häufiges Phänomen, dass bei der Umstellung von einem Referenzarzneimittel auf ein geeignetes Biosimilar oder bei der Umstellung von einem Biosimilar auf ein anderes zu beobachten ist, ist nämlich der Nocebo-Effekt. Dieser beschreibt eine negative gesundheitliche Wirkung während einer Pharmakotherapie, in diesem Fall nach Gabe eines (neuen) Biosimilars, ausgelöst durch die negative Erwartungshaltung beim Patienten. Diese negative Erwartungshaltung kann dadurch entstehen, weil der Patient emotional nur sehr schlecht mit dem Wechsel des Präparats zurecht kommt, weil eine entsprechende heilberufliche Aufklärung fehlte, nur unzureichend war oder gerade zu einer erhöhten Sensibilität geführt hat.
Genau an dieser Stelle liegt einer der größten Kritikpunkte im Hinblick auf den Austausch der Biologika. Eine automatische Substitution im Sinne einer Aut-idem-Regelung, wie bei den klassischen chemisch-synthetischen Generika in Rabattverträgen, verbietet sich, weil gerade im Hinblick onkologischer und chronischer Erkrankungen die Akzeptanz und Adhärenz bei den Patienten in Gefahr sein könnte. Durch die Substitution kann eine negative bis ablehnende Haltung ausgelöst werden, die im Rahmen des Nocebo-Effektes zu unerwünschten Wirkungen oder sogar zu Therapieabbrüchen führen kann. Dies gilt es unbedingt zu verhindern.
Wirkstoff | Referenzarzneimittel | Biosimilars |
|---|---|---|
Rheumatologie, Dermatologie und Gastroenterologie | ||
Adalimumab | Humira® | Amgevita®, Amsparity®, Hyrimoz®, Hulio®, Idacio®, Imraldi® |
Etanercept | Enbrel® | Benepali®, Erelzi®, Nepexto® |
Infliximab | Remicade® | Flixabi®, Inflectra®, Remsima®, Zessly® |
Rituximab | MabThera® | Blitzima®, Ritemvia®, Rixathon®, Riximyo®, Truxima® |
Endokrinologie | ||
Insulin aspart | NovoRapid® | Insulin aspart Sanofi® |
Insulin glargin | Lantus® | Abasaglar®, Semglee® |
Insulin lispro | Humalog® | Insulin lispro Sanofi® |
Teriparatid | Forsteo® | Livogiva®, Movymia®, Terrosa® |
Nephrologie und Onkologie | ||
Epoetin alfa | Eprex® / Erypo® | Anseamed®, Binocrit®, Epoetin alfa Hexal® |
Epoetin zeta | Eprex® / Erypo® | Retactit®, Silapo® |
Onkologie | ||
Bevacizumab | Avastin® | Aybintio®, Equidacent®, Mvasi®, Zirabev® |
Filgrastim | Neupogen® | Accofil®, Filgrastim Hexal®, Grastofil®, Nivestim®, Tevagrastim®, Ratiograstim®, Zarzio® |
Pegfilgrastim | Neulasta® | Cegfila®, Fulphila®, Grasustek®, Nyvepria®, Pelgraz®, Pelmeg®, Udencya®, Ziextenzo® |
Rituximab | MabThera® | Blitzima®, Ritemvia®, Rixathon®, Riximyo®, Truxima® |
Trastuzumab | Herceptin® | Herzuma®, Kanjinti®, Ontruzant®, Ogivri®, Trazimera®, Zercepac® |
Andere Indikationsgebiete | ||
Enoxaparin natrium | Clexane® | Inhixa® |
Enoxaparin natrium | Dezentrales Zulassungsverfahren, Kennzeichnung als Biosimilar | Crusia®, Enoxaparin becat®, Enoxaparin Ledraxen®, Hepaxane® |
Der heilberuflich initiierte Switch
Vielmehr muss die Umstellung von Referenzarzneimitteln auf Biosimilars oder auch beim Austausch von Biosimilars heilberuflich initiiert bzw. begleitet werden. Dieser Vorgang wird als Switch oder auch Transition bezeichnet. Die ärztlich verordnete Umstellung von einem Referenzarzneimittel auf ein Biosimilar, also der Switch, war in den vergangenen Jahren bereits Gegenstand zahlreicher Cross-over-Studien. Auf Grundlage dieser Daten ergeben sich prinzipiell keine Bedenken gegen einen Switch. In zahlreichen klinischen Studien zeigten sich keine (signifikanten) Unterschiede hinsichtlich der Wirksamkeit oder Sicherheit zwischen Referenzarzneimitteln und Biosimilars. So lieferten z. B. die EGALITY-Studie (für Etanercept) und die ADACCESS-Studie (für Adalimumab) keine Hinweise drauf, dass sich eine ärztlich initiierte und begleitete Umstellung der Patienten (sogar in mehrfacher Weise) negativ auf die Therapie mit diesen Biologika auswirken könnte. Aus den bisherigen Studien ableitend ergeben sich keine Bedenken, weshalb eine Umstellung bzw. ein Austausch nicht auf heilberufliche Initiative stattfinden soll – solange beide Seiten, also Ärzte und Apotheker, einbezogen werden und aktiv mitwirken können.
Das Problem der leeren (Kranken-)Kassen
Spätestens nach dem Desaster in der Nachsorgebehandlung von Brustkrebs mit dem Wirkstoff Tamoxifen und den anhaltenden Lieferengpässen, dürfte klar sein, dass die veraltete Logik der Rabattverträge ethisch für derart wichtige Indikationsfelder nicht mehr infrage kommen darf. Die Beobachtungen nach knapp zwei Dekaden Rabattverträgen zeigen: der enorme Preisdruck führt zunehmend zum Outsourcing von großen Teilen der Arzneimittelproduktion in Billiglohnländer, mit denen sich die Europäische Union bereits heute im Wirtschaftskrieg befindet. Zudem sind Effekte im Markt zu beobachten, die sowohl bei den Lieferanten von Rohstoffen, Vor- und Zwischenprodukten, wie auch den pharmazeutischen Unternehmern zu einer Konsolidierung führen. Das Endresultat ist die vollständige Versorgungsabhängigkeit von einzelnen Billigproduzenten in Fernost, die zudem kaum noch kontrollierbar sind. Übertragen auf Biosimilars wäre dies ethisch in keiner Weise vertretbar.
Nicht ganz unproblematisch: Bioidenticals
Bioidenticals oder Reimporte, die aus derselben Produktionsstätte wie das Referenzarzneimittel stammen, können als vollkommen identisch angesehen werden. Den eigentlich bedenkenlosen Austausch erschweren aber die immer noch mangelhaften Informationen nach deutschem Arzneimittelrecht. Zwar sind die pharmazeutischen Unternehmer aus den Fachinformationen ersichtlich, doch die Hersteller sind den Fachkreisen nicht offengelegt, dem Patienten aber sehr wohl, durch die Angaben in der Packungsbeilage. Aus diesem Grund sollte eine finale Begutachtung durch den Gemeinsamen Bundesausschuss (GBA) erfolgen, um Ärzten und Apothekern an dieser Stelle mehr Informationen und Sicherheit zu geben. So wird beispielsweise Interferon beta-1b von den beiden pharmazeutischen Unternehmern Bayer Vital (Betaferon®) und Novartis Pharma (Extavia®) vermarktet, es handelt sich aber um Bioidenticals, da das Produkt mit demselben Prozess am selben Standort produziert wird. Bereits diese Umstellung könnte auf den Patienten befremdlich wirken und die Adhärenz kritisch beeinflussen.
Den Switch kooperativ bewältigen
Befremdlich wirken die Diskussionen, welche schon fast in die sprichwörtlichen Schlammschlachten ausarten, zu Themen wie pharmazeutischen Dienstleistungen oder dem ärztlichen Dispensierrecht für COVID-19-Arzneimittel. Die GKV steht nach rund zweieinhalb Jahren Pandemie finanziell arg gebeutelt da und Wirtschaftlichkeitsreserven müssen in unser aller Interesse realisiert werden.
Ein Switch auf einen preisgünstigeres Bioidentical oder Biosimilar kann ein Beitrag zur Kosteneinsparung sein, den die ärztlichen und apothekerlichen Berufsstände aber nur kooperativ und vor allem gemeinsam mit den Patienten erbringen können. Die Apotheker bringen ihre Marktkenntnis und pharmazeutische Expertise ein. Wichtig ist vor allem die Berücksichtigung aktueller und zukünftig zu erwartender Lieferbarkeiten sowie die Anwendungsfreundlichkeit eingesetzter Applikationssysteme und deren Lagerstabilität. Die Ärzte sind für den diagnostischen Teil der Therapie zuständig und müssen beurteilen, ob eventuell Nocebo-Effekte auftreten und die Therapieadhärenz sowie Heilungschance gefährdet sein könnten.
Wie sich aus den beschriebenen Ausführungen zeigt, bestehen aus regulatorischer und wissenschaftlicher Sicht keine Bedenken für einen heilberuflich initiierten und begleiteten Switch, wohl aber für eine stur nach geltenden Rabatt- bzw. Versorgungsverträgen vollzogene Substitution.
Allerdings muss für die Etablierung eines von Ärzten und Apothekern gemeinsam vollzogenen Biosimilar-Switches noch ein sinnvolles und kooperatives Konzept erarbeitet werden. Berufsverbände, Hersteller, der GBA und nicht zuletzt die Verantwortlichen in der Gesundheitspolitik haben bis zum 16. August 2023 nun noch Zeit. Dieser Aufschub macht also Sinn und sollte daher sinnvoll genutzt werden. |
Literatur
Arzneimittelkommission der deutschen Ärzteschaft: Biosimilars – Leitfaden der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ), 2. Auflage, Version 1.0, Januar 2021
Blauvelt A, Lacour JP, Fowler JF, Jr. et al.: Phase III rando-mized study of the proposed adalimumab biosimilar
GP2017 in psoriasis: impact of multiple switches. Br J Dermatol 2018; 179: 623-631
Dicheva-Radev, S: Potenziale und Risiken von Biologika und Biosimilars: Empfehlungen der Arzneimittelkommission der deutschen Ärzteschaft zum Einsatz von Biosimilars. Vortrag am 7. März 2020 in Bremen
Europäischen Arzneimittel-Agentur, Europäischen Kommission: Biosimilars in der EU, Leitfaden vom 2. Oktober 2019
Griffiths CEM, Thaci D, Gerdes S et al.: The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol 2017;176:928-938
Autor
Dr. Uwe Weidenauer, Apotheker, Studium in Heidelberg, Promotion in Pharmazeutischer Technologie in Marburg, schlug danach eine Industrielaufbahn ein und arbeitete in verschiedenen Positionen in Forschung und Entwicklung sowie als Herstellungsleiter. Nach einigen Jahren in verschiedenen Managementpositionen entschloss er sich, zurück zu seinen Wurzeln zu kehren und mehrere Apotheken zu übernehmen.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.