














- DAZ.online
- DAZ / AZ
- DAZ 34/2014
- Arzneimittelzulassung

nmann77 - Fotalia.com
Arzneimittelrecht
Arzneimittelzulassung
Teil 1: Nationale Zulassung von neuen und bekannten Stoffen, Zulassung von Generika
Die Zulassung von Arzneimitteln ist Gegenstand des vierten Abschnitts des Arzneimittelgesetzes mit den §§ 21 bis 37. Weitere Einzelheiten können per Rechtsverordnung festgelegt werden (§35AMG). Zusätzlich veröffentlichen die Bundesoberbehörden in loser Folge in Bekanntmachungen im Bundesanzeiger interne Verfahrensregeln hierzu.
Die für die Arzneimittelzulassung zuständigen Bundesoberbehörden sind
- das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM),
- das Paul-Ehrlich-Institut (PEI) für Sera, Impfstoffe, Blutzubereitungen, Knochenmarkzubereitungen, Gewebezubereitungen, Gewebe, Allergene, Arzneimittel für neuartige Therapien, xenogene Arzneimittel und gentechnisch hergestellte Blutbestandteile und
- das Bundesamt für Verbraucherschutz und Lebensmittelsicherheit (BVL) für Tierarzneimittel.
Zulassungspflicht
Welche Arzneimittel zulassungspflichtig sind, legt § 21 AMG fest. Im Wesentlichen sind dies Fertigarzneimittel (die Arzneimittel im Sinne des § 2 Abs. 1 oder Abs. 2 Nr. 1 sind, siehe Folge zum Arzneimittelbegriff in der DAZ Nr. 18, 2014, S.58–62). Von der Zulassungspflicht ausgenommen sind z.B. Rezepturarzneimittel. Auch Humanarzneimittel, die aufgrund nachweislich häufiger ärztlicher Verschreibung in den wesentlichen Herstellungsschritten in einer Apotheke in Chargengrössen bis zu hundert abgabefertigen Packungen pro Tag im Rahmen des üblichen Apothekenbetriebs hergestellt und zur Abgabe in dieser Apotheke bestimmt sind, bedürfen keiner Zulassung (sogenannte „Hunderter-Regel“). Des Weiteren unterliegen Arzneimittel, die zur klinischen Prüfung bestimmt sind, nicht der Zulassungspflicht.
Formale Voraussetzungen für die Zulassung
Die Zulassung muss vom pharmazeutischen Unternehmer beantragt werden. Für ein Fertigarzneimittel, das in Apotheken aufgrund einheitlicher Vorschriften hergestellt und unter einer einheitlichen Bezeichnung an Verbraucher abgegeben wird, muss der Herausgeber der Herstellungsvorschrift die Zulassung beantragen. Bei der Antragstellung muss angegeben werden, ob eine Zulassung des Arzneimittels in anderen Staaten beantragt, erteilt, versagt oder ein solcher Antrag zurückgenommen wurde. Besonders wichtig sind Angaben zu anderen Zulassungen und Verfahren in weiteren EU-Mitgliedstaaten, weil in diesen Fällen besondere Modalitäten im Rahmen des EU-Zulassungssystems zu beachten sind.
Formale Anforderungen an die Zulassungsunterlagen
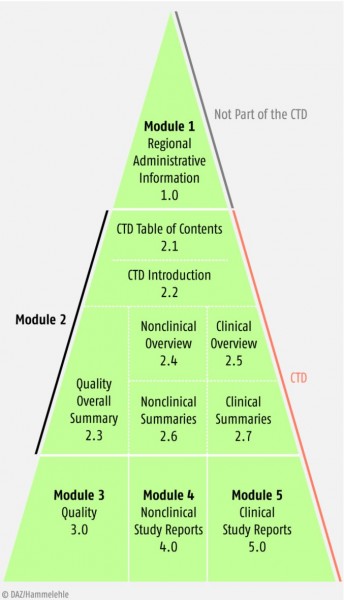
Hinsichtlich der formalen Anforderungen an die Zulassungsunterlagen wird heute im Wesentlichen auf europäische bzw. international harmonisierte Vorgaben zurückgegriffen. Das maßgebliche Dokument ist die sogenannte „Notice to Applicants“ (NtA, Band 2 für Humanarzneimittel des europäischen Regelwerks für Arzneimittel EudraLex). Die NtA inkorporiert die international im Rahmen der International Conference on Harmonisation (ICH) zwischen der EU, USA und Japan harmonisierten Vorgaben des sogenannte Common Technical Documents (CTD). Hiernach ist ein Zulassungsantrag modular aufgebaut (siehe Abb. 1).
[CTD = Common Technical Documents]
Modul 1 enthält administrative Dokumente wie den Antrag selbst und die informativen Texte (SPC, Packungsbeilage, Kennzeichnung), Modul 2 die Quality Overall Summary (2.3), die bewertenden Overviews zur Präklinik und Klinik (2.4 und 2.5) sowie die zusammenfassenden Übersichten zu den präklinischen und klinischen Ergebnissen (2.6 und 2.7). In den Modulen 3, 4 und 5 sind schließlich alle ausführlichen Dokumente zu den Studien und/oder auch bibliografische Unterlagen enthalten.
Seit August 2013 verzichtet das BfArM für alle Antragsverfahren bei Nutzung des PharmNet.Bund-Portals oder des europäischen Portals CESP sowie bei Nutzung anderer elektronischer Speichermedien (CD/DVD) auf eine Einreichung in Papierform.
Zulassung von neuen Stoffen
Die inhaltlichen Anforderungen an die Zulassungsunterlagen für neue Arzneimittel mit eigenen Studien (stand alone application) sind in § 22 bis 24 AMG festgelegt. Wesentlichster Teil des Antrags sind die Ergebnisse der analytischen, der pharmakologisch-toxikologischen und der klinischen Prüfung des Arzneimittels. Dem Antrag müssen alle für die Bewertung zweckdienlichen Angaben und Unterlagen beigefügt werden, ob günstig oder ungünstig. Für besondere Arzneimittelgruppen werden noch weitere Unterlagen gefordert, so zum Beispiel bei Kombinationsarzneimitteln eine Begründung, dass jeder arzneilich wirksame Bestandteil einen Beitrag zur positiven Beurteilung des Arzneimittels leistet (§ 22 Abs. 3a AMG). Außerdem müssen für bestimmte Stoffe und Zubereitungen Unterlagen zur Bioverfügbarkeit bzw. im Falle einer bezugnehmenden Zulassung zur Bioäquivalenz vorgelegt werden.
Zulassungsverfahren
Das Zulassungsverfahren gliedert sich in zwei Phasen:
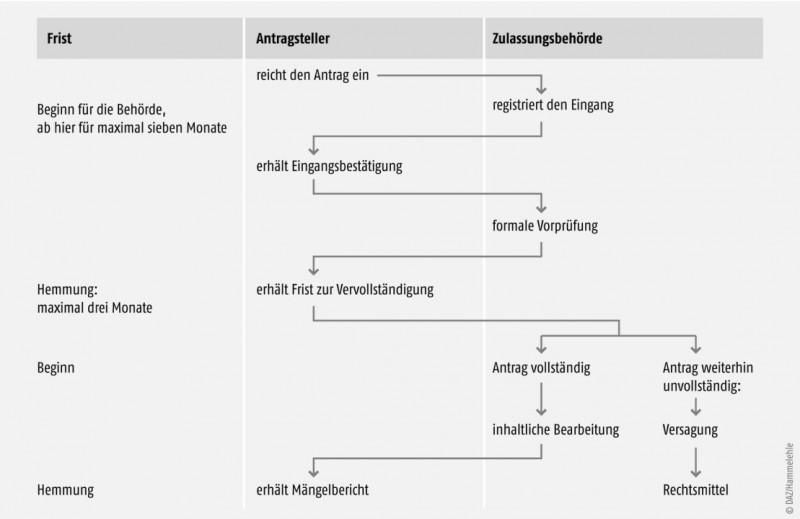
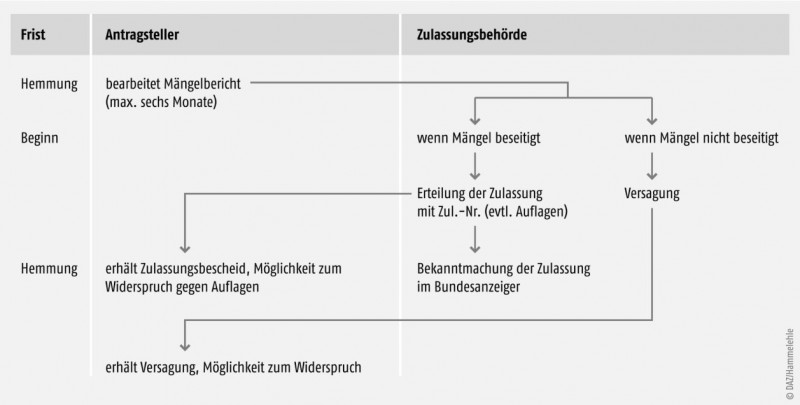
- Phase 1 ist die erste Überprüfung der Unterlagen, die mit dem Mängelbericht – soweit Mängel festgestellt werden – abschließt.
- Phase 2 umfasst die Beurteilung der Beantwortung durch den Antragsteller bis zum abschließenden Bescheid (siehe Abb. 2 und 3).
Antragseingang und Fristen für die Bearbeitung
Nachdem der Antrag bei der Zulassungsbehörde eingegangen ist, erhält er eine Bearbeitungsnummer, und der Antragsteller bekommt eine Eingangsbestätigung (Abb. 2). Damit beginnt die Bearbeitungsfrist von sieben Monaten für die Behörde zu laufen (§ 27 AMG). Bei der Berechnung ist zu beachten, dass die Frist immer dann gehemmt wird, wenn der Antragsteller Gelegenheit zur Behebung von Mängeln erhält.
Wie prüft die Zulassungsbehörde?
Als ersten Prüfschritt kann die Behörde den Zulassungsantrag formal vorprüfen lassen (Abb. 2). Ist diese Hürde genommen, so steigt sie in die inhaltliche Bearbeitung ein. Handlungsanweisung hierfür sind die Arzneimittelprüfrichtlinien, deren Inhalte heute weitgehend auf europäischen Vorgaben beruhen (siehe auch vorherige Folgen der DAZ-Rechtsserie). Bei der Beurteilung ist die ausschlaggebende Frage letztendlich, ob das Nutzen-Risiko-Verhältnis (§ 4 Abs. 28 AMG) positiv ist.
Bei Beanstandungen der Unterlagen erhält der Antrag steller die Gelegenheit, den Mängeln innerhalb einer angemessenen Frist abzuhelfen (Mängelbericht gemäß §25Abs. 4AMG). Die Frist darf sechs Monate nicht überschreiten (Abb. 3).
Zulassungsversagungsgründe
Das Gesetz geht für den „Normalfall“ davon aus, dass die Zulassung erteilt wird. Nicht umsonst lautet die Formulierung in § 25 Abs. 2 AMG „die zuständige Bundesoberbehörde darf die Zulassung nur versagen, wenn...“. Genannt wird im Folgenden ein abschließender Katalog von Tatbeständen (siehe Kasten „Zulassungsversagungsgründe“), von denen allerdings bereits einer für die Versagung der Zulassung ausreicht.
Zulassungsversagungsgründe nach § 25 AMG
- Unterlagen unvollständig
- Arzneimittel nicht ausreichend geprüft
- Qualität nicht ausreichend
- therapeutische Wirksamkeit fehlt oder ist unzureichend begründet
- ungünstiges Nutzen-Risiko-Verhältnis
- Kombinationsbegründung unzureichend
- Inverkehrbringen des Arzneimittels würde gegen gesetzliche Vorschriften oder gegen eine EU/EG-Verordnung oder EU/EG-Richtlinie verstoßen
- entsprechende Standardzulassung vorhanden
Erteilung der Zulassung
Die zuständige Bundesoberbehörde erteilt die Zulassung schriftlich unter Zuteilung einer Zulassungsnummer. Der Antragsteller muss für das Verfahren eine Gebühr entrichten, die für neue Stoffe nach der AMG-Kostenverordnung gegenwärtig bei rund 51.000 Euro liegt. Verschiedene Darreichungsformen eines Arzneimittels gleicher Bezeichnung oder verschiedene Konzentrationen eines Arzneimittels gleicher Darreichungsform können eine einheitliche Zulassungsnummer erhalten. Zur Unterscheidung werden weitere Kennzeichen hinzugefügt (§ 25 Abs. 9 AMG). Erst mit dem Erhalt des Zulassungsbescheides wird das Arzneimittel verkehrsfähig. Zur Information der Fachkreise wird die Erteilung oder Verlängerung der Zulassung wie auch deren Rücknahme oder Erlöschen im Bundesanzeiger bekannt gemacht. Wichtig: Die Zulassung lässt die zivil- und strafrechtliche Verantwortlichkeit des pharmazeutischen Unternehmers für das in Verkehr gebrachte Arzneimittel unberührt (§ 25 Abs. 10 AMG).
Zulassung mit Auflagen
Die Behörde kann die Zulassung mit Auflagen z.B. bezüglich der Kennzeichnung oder des Wortlauts der Packungsbeilage und der Fachinformation versehen (§ 28 AMG). Auflagen können sich auch auf Packungsgrößen oder Behältnisse beziehen oder die Anwendung des Arzneimittels auf Ärzte bestimmter Fachgebiete oder auf Spezialkliniken beschränken. Sie können im Übrigen auch nach der erteilten Zulassung jederzeit angeordnet werden. So können zum Beispiel auch Wirksamkeits- und Sicherheitsstudien (post-authorisation efficacy studies/PAES bzw. post-authorisation safety studies/PASS) unter besonderen Voraussetzungen per Auflage nachgefordert werden (§ 28 Abs. 3b AMG).
Zulassung von bekannten Stoffen
Anstelle der Ergebnisse der pharmakologisch-toxikologischen und klinischen Prüfung kann für bekannte Stoffe sogenanntes „anderes wissenschaftliches Erkenntnismaterial“ vorgelegt werden (§ 22 Abs. 3 AMG), und zwar für
- Arzneimittel mit Wirkstoffen, die in der EU seit mindestens zehn Jahren allgemein medizinisch verwendet wurden (well-established-use), deren Wirkungen und Nebenwirkungen bekannt und aus dem wissenschaftlichen Erkenntnismaterial ersichtlich sind,
- Arzneimittel, die mit diesen vergleichbar sind,
- neue Kombinationen bekannter Wirkstoffe, entweder für diese selbst oder auch für die Kombination.
Mit anderem wissenschaftlichem Erkenntnismaterial sind zum Beispiel allgemeine wissenschaftliche Literatur zu dem Wirkstoff oder publizierte Studien mit Vergleichspräparaten gemeint. Ein solches Genehmigungsverfahren führt zu einer bibliografischen (well-established-use/WEU) Zulassung.
Zulassungsverlängerung
Mit der Zulassung wird nicht automatisch die Genehmigung erteilt, das Arzneimittel zeitlich unbegrenzt in den Verkehr zu bringen. Fünf Jahre nach Erteilung der Zulassung muss einmalig ein Antrag auf Verlängerung gestellt werden (renewal). Der Zulassungsinhaber muss hierzu einen Bericht über die bisherigen Erfahrungen mit dem Arzneimittel einreichen. Wird der Antrag positiv beschieden, so gilt die Zulassung danach unbegrenzt fort. In begründeten Fällen, wie etwa aus Arzneimittelsicherheitsgründen, kann eine weitere befristete Verlängerung angeordnet werden.
Erlöschen der Zulassung und sunset clause
Für den Fall, dass ein Unternehmen keinen Verlängerungsantrag stellt oder selbst freiwillig auf eine bestehende Zulassung verzichtet, darf das Arzneimittel noch für einen begrenzten Zeitraum weiter vertrieben werden. Seit der letzten Novellierung des AMG müssen der Zulassungsbehörde allerdings unverzüglich die Gründe für solche Entscheidungen angezeigt werden. Außerdem müssen die Zulassungsinhaber bei der Behörde Meldung erstatten, wenn sie das Arzneimittel in den Verkehr bringen oder wenn das Inverkehrbringen vorübergehend oder endgültig eingestellt wird (§ 29 Abs. 1b und 1c AMG). Hierzu haben die Behörden spezielle online-Portale eingerichtet. Wird ein Arzneimittel innerhalb von drei Jahren nach Erteilung der Zulassung nicht in den Verkehr gebracht wird oder befindet es sich nach der Zulassung in drei aufeinander folgenden Jahren nicht mehr im Verkehr, so erlischt die Zulassung (sunset clause, § 31 Abs.1 Nr. 1 AMG).
Zulassung von Generika
Laufen Schutzfristen für ein neues Arzneimittel aus, so stehen vor allem dann, wenn es sich um ein hochpreisiges, umsatzstarkes Produkt handelt, die Nachahmer bereits in den Startlöchern. Sie werden in der Regel versuchen, eine Zulassung unter Bezugnahme auf die Unterlagen des Erstanbieters zu beantragen. Bei Generika-Zulassungen ist die Übereinstimmung mit dem Referenzarzneimittel von großer Bedeutung. Je nach Umfang der Abweichung ist eine Bezugnahme entweder nicht möglich, oder es müssen ergänzende Unterlagen vorgelegt werden.
Definition Generikum
„im Wesentlichen gleich“ („essentially similar“) zum Referenzarzneimittel
Das bedeutet:
- gleicher Wirkstoff nach Art und Menge wie das Referenzarzneimittel,
- gleiche Darreichungsform,
- Nachweis der Bioäquivalenz mit dem Referenzarzneimittel.
Erlaubte Abweichungen:
- Wirkstoff: verschiedene Salze, Ester, Ether, Isomere etc., außer ihre Eigenschaften unterscheiden sich erheblich hinsichtlich Unbedenklichkeit oder Wirksamkeit
- Darreichungsform: verschiedene schnellfreisetzende orale Darreichungsformen werden als gleich eingestuft
- Bioäquivalenzstudien: in bestimmten Fällen verzichtbar (Biowaiver)
Quelle: § 24b Abs. 2 AMG und CHMP Guideline on the Investigation of Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1/ Corr)
Referenzarzneimittel und Schutzfrist
Die Unterlagenschutzfrist wird gerechnet ab dem Zeitpunkt der Erstzulassung des Referenzarzneimittels. Durch die Einbindung Deutschlands in die EU wird jedoch nicht die Erstzulassung in Deutschland, sondern in irgendeinem Mitgliedstaat als Ausgangspunkt genommen (§ 24b Abs. 1 AMG). Dabei muss es sich nicht unbedingt um ein vollkommen identisches Arzneimittel handeln, denn bei der Bezugnahme gilt das Prinzip der „global marketing authorisation“ (siehe Kasten). Die erste Zulassung löst die Schutzfrist aus, und wenn diese abgelaufen ist, sind auch alle nachfolgenden Einzelzulassungen, etwa für weitere Stärken und Darreichungsformen nicht mehr geschützt.
Prinzip der global marketing authorisation
Für Zulassungen nach § 24b Abs. 1 (Generika-Zulassungen) gelten Einzelzulassungen eines Referenzarzneimittels als einheitliche umfassende Zulassung.
Quelle: § 25 Abs. 9 AMG
Bezugnahme mit und ohne Zustimmung
Grundsätzlich kann ein Antragsteller jederzeit auf die noch geschützten Unterlagen eines Vorantragstellers Bezug nehmen, wenn er hierzu dessen schriftliche Zustimmung vorlegen kann (§ 24a AMG, informed consent). Der Umfang der möglichen Bezugnahme ist dann sogar noch weitergehend als nach Ablauf der Unterlagenschutzfrist und umfasst auch den Teil zur Qualität. Kann der Zweitanmelder nachweisen, dass das Bezugspräparat in einem Mitgliedstaat der EU bereits seit mindestens acht Jahren zugelassen ist oder vor mindestens acht Jahren zugelassen wurde, so ist die Schutzfrist abgelaufen (acht Jahre Unterlagenschutz) und er braucht für die Bezugnahme keine Zustimmung des Vorantragstellers mehr (§ 24b AMG). Er kann dann allerdings im Wesentlichen nur auf die Ergebnisse zur Pharmtox und Klinik und die Sachverständigengutachten Bezug nehmen. Die Bezugnahme ist auch möglich, wenn die Referenzzulassung gar nicht mehr besteht. Der Antrag kann in der Behörde direkt bearbeitet werden.
Nicht verwechseln! Unterlagen-, Vermarktungs- und Patentschutz
Auch wenn die Nachahmer-Zulassung bereits erteilt ist, darf ein Generikum frühestens nach Ablauf von zehn Jahren nach Erteilung der ersten Genehmigung für das Referenzarzneimittel in den Verkehr gebracht werden (zehn Jahre Vermarktungsschutz). Der Schutzzeitraum des Originators für die Vermarktung kann auf höchstens elf Jahre verlängert werden, wenn er innerhalb von acht Jahren nach Erstzulassung die Erweiterung um eines oder mehrere neue Anwendungsgebiete von bedeutendem klinischem Nutzen im Vergleich zu bestehenden Therapien erwirkt.
Neben den Schutzrechten des Innovators, die sich aus dem Arzneimittelrecht ergeben, greifen für den Vermarktungsschutz auch noch patenrechtliche Regelungen, wie etwa Wirkstoff-, Verfahrens- oder Nutzer-Patente, auf die hier nicht näher eingegangen werden soll. Wichtig ist, dass Wirkstoff-Patente (Laufzeit: 20 Jahre) in der Regel sehr früh beantragt werden und bei den langen Entwicklungszeiten eines Arzneimittels vielfach schon fast abgelaufen sind, bevor die Innovation auf den Markt kommt. Deswegen kann gegebenenfalls ein ergänzendes Schutzzertifikat (Supplementary Protection Certificate, SPC) beantragt werden, dass die Laufzeit des Grundpatentes maximal um fünf Jahre verlängert.
Standardzulassung
Die Standardzulassung (§ 36 AMG) ist eine deutsche Besonderheit mit einer speziellen Vorgeschichte. Hiermit können bestimmte Arzneimittel oder Arzneimittelgruppen von der Pflicht zur Einzelzulassung freigestellt werden. Die Standardzulassungstexte werden in Anlehnung an „normale“ Zulassungen von einem eigens zu diesem Zweck eingerichteten Sachverständigenausschuss beim BfArM erarbeitet. Standardzulassungen werden ihrer Zweckbestimmung entsprechend gerne für die Arzneimittelherstellung in Krankenhausapotheken und öffentlichen Apotheken genutzt. Ihre Nutzung muss der Zulassungs- und der zuständigen Länderbehörde angezeigt werden (§ 67 Abs. 5 AMG).
Mammutaufgabe Nachzulassung
Obwohl die Nachzulassung mittlerweile schon fast Geschichte ist, sollen ihr doch einige Ausführungen gewidmet werden, denn sie hat letzten Endes zu einer tiefgreifenden Bereinigung des deutschen Arzneimittelmarktes geführt. Nach der nationalen Umsetzung der pharmazeutischen Basisrichtlinien (siehe Folge 2 der DAZ-Serie in der DAZ 2014, Nr. 14, S. 54) hatte die EU für die Überprüfung der Bestandsmärkte in den Mitgliedstaaten auf Qualität, Wirksamkeit und Unbedenklichkeit eine Frist bis zum 20. Mai 1990 vorgegeben. In Deutschland erhielten die Präparate zunächst eine „fiktive Zulassung“, die sie berechtigte, bis zum Ablauf der Übergangsfrist weiter in Verkehr zu bleiben.
Im Jahr 1978 waren zunächst 140.000 Arzneimittel beim damaligen BGA gemeldet worden (Stärken und Darreichungsformen einzeln gezählt), eine schier unübersehbare Menge an Präparaten. Bis zum Jahr 1990 hatte sich diese Zahl durch Löschungen auf 126.000 Arzneimittel reduziert. Hinzu kamen jedoch ca. 3800 Arzneimittel mit einer fiktiven Zulassung aus den neuen Bundesländern im Zuge der Wiedervereinigung. Tatsächlich gingen dann insgesamt 61.000 Anträge ins Nachzulassungs- bzw. Registrierungsverfahren (homöopathische Arzneimittel).
Die Aufbereitung und ihr Ende
Die Präparate-spezifische Überprüfung des „Altarzneimittelmarktes“ ließ einen immensen Prüfaufwand erwarten. Tatsächlich waren in den fiktiv zugelassenen Arzneimitteln jedoch etwa „nur“ 2500 chemisch definierte Stoffe und rund 1000 verschiedene Heilpflanzen enthalten. So versprach sich der Gesetzgeber seinerzeit eine erhebliche Rationalisierung des Verfahrens davon, das vorliegende wissenschaftliche Erkenntnismaterial zu den Wirkstoffen von speziellen Kommissionen in Monografien aufbereiten zu lassen. Auf Basis dieser Aufbereitungsmonografien sollten die Fertigarzneimittel dann im Rahmen der Nachzulassung beurteilt werden. Insgesamt fünfzehn Aufbereitungskommissionen wurden zu diesem Zweck eingerichtet. Auf den ersten Blick als Ideallösung angesehen, waren die Arbeiten an den Monografien zwar produktiv, gingen jedoch insgesamt viel zu langsam voran. So wurde die Aufbereitung schließlich im Jahr 1994 wieder eingestellt. Während der Prozess in den meisten Ländern zügig voranging, waren in Deutschland bis dahin erst zehn Nachzulassungen erteilt worden. Die Nachzulassung hatte sich mittlerweile zu einem echten „Sorgenkind“ der deutschen Gesundheitspolitiker in den neunziger Jahren entwickelt.
Die Ex-ante-Unterlagen
In Verbindung mit der Einstellung der Aufbereitung wurde den Antragstellern die Beweislast für die Wirksamkeit und Unbedenklichkeit der Altarzneimittel aufgebürdet. Dies bedeutete zwar nicht, dass sie nun automatisch individuell klinische und toxikologische Studien für ihre Präparate vorlegen mussten. Sie konnten sich hierzu im Wesentlichen weiterhin auf die Monografien und auf Literaturdaten (bibliografische Daten) berufen, mussten diese aber in aufbereiteter Form als so genannte „Ex-ante-Unterlagen“ zur Beurteilung beim BfArM einreichen – ein riesiger Kraftakt. In jahrelanger, mühevoller „Kleinarbeit“ konnte der Berg in den Zulassungsbehörden schließlich abgearbeitet werden.
Was heute vom so genannten Altarzneimittelmarkt übrig geblieben ist, zeigt Tab. 1 mit dem aktuellen Stand zum deutschen Arzneimittelmarkt. Hiernach haben lediglich rund 5500 Arzneimittel die Hürde der Nachzulassung und ca. 2600 Homöopathika und Anthroposophika die der Nachregistrierung genommen.
Autorin
Dr. Helga Blasius, Fachapothekerin für Arzneimittelinformation, Dipl.-Übersetzerin (Jap., Kor.)



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.