











- DAZ.online
- DAZ / AZ
- DAZ 38/2014
- Arzneimittelzulassung
nmann77 - Fotalia.com
Arzneimittelzulassung
Teil 2: Europäisches Zulassungssystem, Änderungen an Arzneimitteln nach der Zulassung
Die europäische Zulassungsagentur
Die European Medicines Agency (EMA, früher EMEA), die ihre Tätigkeit am 1. Januar 1995 aufgenommen hat, ist eine der wichtigsten Säulen des gemeinsamen Binnenmarktes für Arzneimittel. Sie dient als wissenschaftliche Beratungsstelle und macht innovative Arzneimittel schneller für die Patienten verfügbar. Außerdem koordiniert sie die einzelstaatlichen Maßnahmen auf dem Gebiet der Arzneimittelsicherheit und der Überwachung unter den Mitgliedstaaten. Mit der Übertragung dieser Kompetenzen wird die Rolle der nationalen Behörden jedoch keineswegs geschmälert. Sie bilden vielmehr die Grundpfeiler für die Arbeit der EMA und sind nach wie vor verantwortlich für die rein nationalen Märkte in den Mitgliedstaaten.
Die Agentur hat sieben wissenschaftliche Ausschüsse (Tab. 1), ein Sekretariat, einen Verwaltungsdirektor (Executive Director, derzeit der Italiener Guido Rasi) und einen Verwaltungsrat (Management Board). Die Ausschüsse setzen sich aus Vertretern der nationalen Zulassungsbehörden der Mitgliedstaaten zusammen. Sie werden jeweils durch eigene ständige und nicht ständige Arbeitsgruppen unterstützt. Darüber hinaus kann die Agentur auf einen umfangreichen Fundus an externen Gutachtern zurückgreifen, die in einem europäischen Expertenregister erfasst sind.
Aufgaben der Ausschüsse
Die Ausschüsse, vor allem der CHMP und der CVMP, erfüllen vielfältige Aufgaben. Sie betreuen die zentralen Zulassungsverfahren und fungieren als Schiedsstelle bei Meinungsverschiedenheiten der nationalen Behörden im dezentralen und im Anerkennungsverfahren. Darüber hinaus erarbeiten sie die zahlreichen Leitlinien und Empfehlungen (Guidelines und Notes for Guidance), die bei der Prüfung und Zulassung bzw. Registrierung von Arzneimitteln zu beachten sind. Die Codierung einer wissenschaftlichen Guideline gibt Aufschluss darüber, aus welchem Fachgremium sie stammt. „CHMP/EWP/342691/09“ kommt z.B. aus der Arbeitsgruppe Wirksamkeit (Efficacy Working Party) des Ausschusses für Humanarzneimittel. Enthält sie das Kürzel ICH, so wurden die Inhalte im Rahmen der International Conference on Harmonisation (ICH) zwischen der EU, den USA und Japan harmonisiert (z.B. CHMP/ICH/541/00).
Die Ausschüsse kommen in der Regel einmal im Monat für mehrere Tage zusammen (außer im August). Die Termine, Tagesordnungen und Sitzungsprotokolle werden auf der Webseite der EMA bekannt gemacht. Die Arbeit der EMA ist insgesamt sehr transparent.
Das europäische Zulassungssystem
Das Zulassungssystem der Europäischen Union ist am 1. Januar 1995 in Kraft getreten. Es besteht aus dem zentralen Verfahren, dem gegenseitigen Anerkennungsverfahren und dem dezentralisierten Verfahren, das erst im Jahr 2005 eingerichtet wurde (Tab. 2 und Tab. 3).
Zentrales Zulassungsverfahren
Über ein zentrales Zulassungsverfahren kann ein Antragsteller gleichzeitig die Verkehrsgenehmigung in allen Mitgliedstaaten der EU und des europäischen Wirtschaftsraums (d.h. zusätzlich in Island, Liechtenstein und Norwegen) erzielen, eine attraktive Option, aber das Verfahren ist nicht für alle Arzneimittel zugänglich (Tab. 4).
Die Anträge, die sehr gut vorbereitet und vorher bereits mit der EMA abgestimmt werden müssen, werden direkt bei der Agentur in London eingereicht. Für jedes Verfahren werden zwei Behörden als Berichterstatter (Rapporteur) und als Mitberichterstatter (Co-Rapporteur) benannt. Sie erstellen einen Bewertungsbericht (Assessment Report, AR) zu dem Antrag.
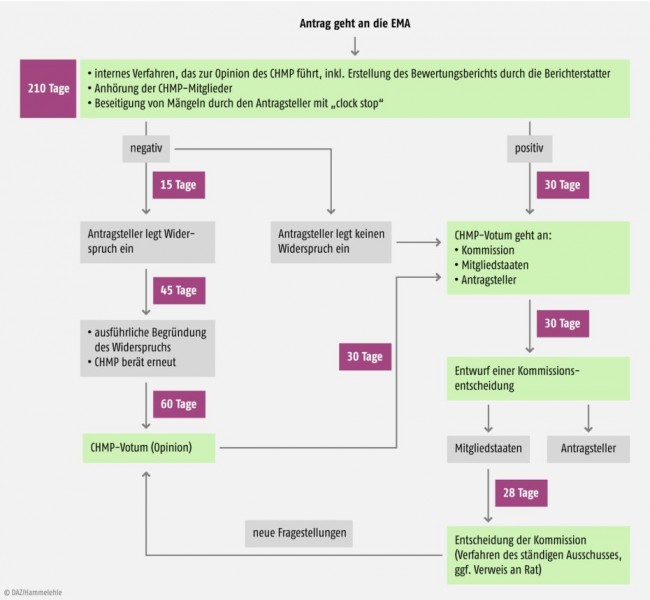
Dieser wird von den übrigen Mitgliedstaaten geprüft und anschließend im CHMP diskutiert und verabschiedet. Bei Mängeln erhält der Antragsteller gegebenenfalls Gelegenheit zur Nachbesserung (list of questions), wodurch die Bearbeitungsfrist für die Behörde gehemmt, d.h. unterbrochen wird (clock stop). Die Bewertung muss innerhalb von 210 Tagen abgeschlossen sein. Sie mündet in ein positives oder negatives Votum (Opinion) für die Zulassungsentscheidung. Die eigentliche Zulassung erteilt nicht die Agentur selbst, sondern anschließend die Europäische Kommission in Form eines „Verwaltungsaktes der EU“ (Binding Community Decision). Zum Ablauf des zentralen Zulassungsverfahrens inklusive Widerspruchsmöglichkeiten siehe Abb. 1.
EPAR, Register, Zulassungsnummer
Nach Erteilung der Zulassung publiziert die EMA einen öffentlichen Bewertungsbericht (European Public Assessment Report, EPAR) zu dem Arzneimittel. Die Zulassung wird im Amtsblatt der Europäischen Union bekanntgemacht und in das Register der zentralen Zulassungen (Community Register) aufgenommen. Zentrale Zulassungen sind daran erkennbar, dass ihre Zulassungsnummer mit „EU“ anfängt.
Eine zentrale Zulassung ist im Regelfall für fünf Jahre gültig und muss dann wie nationale Zulassungen einmalig verlängert werden. Danach gilt sie in der Regel ohne zeitliche Begrenzung. Der Unterlagen- und Vermarktungsschutz für zentral zugelassene Arzneimittel ist derselbe wie bei nationalen Zulassungen.
Grundidee des MRP und DCP
Für Arzneimittel, die nicht unter den Anwendungsbereich des zentralen Verfahrens fallen, muss, wenn sie in mehreren Mitgliedstaaten zur Zulassung gebracht werden sollen, entweder das Verfahren der gegenseitigen Anerkennung oder das dezentrale Verfahren beschritten werden (Tab. 3). Wichtig: In beiden Fällen ist es nicht möglich, gesonderte parallele nationale Zulassungen ohne Abstimmung unter den Behörden zu beantragen, da hiermit die Harmonisierung der nationalen Arzneimittelmärkte innerhalb der EU umgangen würde (§ 25a AMG). Rein nationale Zulassungen sind in der EU nur noch möglich, wenn ein Präparat lediglich in einem Mitgliedstaat auf den Markt gebracht werden soll.
Ablauf des MRP und des DCP
In beiden Verfahren kann der Antragsteller die Länder, in denen er die Zulassung anstrebt, selbst bestimmen. Liegt schon eine Zulassung vor, so dient sie als Basiszulassung für die Anerkennung (MRP), liegt keine Zulassung vor, so wird in den gewünschten Mitgliedstaaten ein Antrag mit einem identischen Dossier eingereicht (DCP).
In Anlehnung an das zentrale Verfahren wird bei beiden Verfahren die Behörde eines Mitgliedstaates als Koordinator (heißt hier: Reference Member State, RMS) bestimmt. Im MRP fungiert die Behörde des Mitgliedstaates, die die Basiszulassung erteilt hat, als RMS. Der RMS erstellt auf der Basis der bereits bestehenden Zulassung (im MRP) bzw. des neu vorgelegten Dossiers (im DCP) einen Bewertungsbericht (im MRP innerhalb von 90 Tagen, im DCP innerhalb von 120 Tagen). Dieser wird den Behörden der anderen beteiligten Mitgliedstaaten (Concerned Member States, CMS) und dem Antragsteller zusammen mit Entwürfen für die Zusammenfassung der Merkmale des Arzneimittels (SPC), Etikettierung und Packungsbeilage zur Überprüfung und Annahme weitergeleitet.
Die CMSs sollen den Bewertungsbericht und die Texte dann innerhalb von 90 Tagen anerkennen und innerhalb weiterer 30 Tage die entsprechenden nationalen Zulassungen erteilen.
Wichtig: Im Unterschied zum zentralen Verfahren resultieren aus dem Anerkennungs- und dem dezentralen Verfahren nationale Zulassungen, die jedoch harmonisiert sind. Um diese Harmonisierung nicht zu gefährden, müssen in der Folge sämtliche Änderungen an dem Arzneimittel zwischen den Mitgliedstaaten abgestimmt und konzertiert umgesetzt werden. Arzneimittel, die nach einem DCP oder MPR zugelassen wurden, leben also nicht national, sondern europäisch „fort“ (s.u.).
Im Streitfall: Schiedsverfahren
Was passiert, wenn ein Mitgliedstaat die Anerkennung verweigert? Bedenken und Ablehnungen sind nur in sehr eingeschränktem Maße möglich, und zwar dann, wenn ein CMS eine „ernsthafte Gefahr für die öffentliche Gesundheit“ (serious risk to public health) durch das Präparat geltend macht. Die Europäische Kommission hat hierzu eigens eine Guideline ausgearbeitet, um nationale „Empfindlichkeiten“ bei der Anerkennung möglichst zu begrenzen. Bei Meinungsverschiedenheiten hilft ein Schlichtungsgremium, die Coordination Group for Mutual Recognition Procedures and Decentralised Procedures (CMD; h für human). Gelingt es der CMD (h) nicht, einen Konsens zu erzielen, wird der Fall an den Arzneimittelausschuss (CHMP) verwiesen (referral), und es kommt zu einem Schiedsverfahren (arbitration procedure). Das Votum des Ausschusses mündet in eine abschließende Entscheidung der Europäischen Kommission, die dann in allen Mitgliedstaaten, in denen das Arzneimittel in Verkehr ist, umgesetzt werden muss. Das kann im schlimmsten Fall auch dazu führen, dass eine bestehende Zulassung wieder zurückgenommen werden muss. Auf den Ablauf des Schiedsverfahrens soll hier nicht näher eingegangen werden.
Datenbanken für Arzneimittelzulassungen in der EU, EMA und HMA
Deutschland www.pharmnet-bund.de/dynamic/de
Zentrale EU-Zulassungen ec.europa.eu/health/documents/community-register
EPARs www.ema.europa.eu/ema → find medicine → human medicines
Zulassungen über MRP und DCP mri.medagencies.org/Human
Andere EU-Mitgliedstaaten ec.europa.eu/health/documents/community-register/regca_en.htm
HMA* www.hma.eu
* Heads of Medicines Agencies, gemeinsame Webseite der nationalen Zulassungsagenturen
Bedingte Zulassung
In Ausnahmefällen können zentrale Zulassungen auch erteilt werden, wenn ein Antragsteller nicht alle erforderlichen Daten vorlegen kann. So kann zum Beispiel für Arzneimittel zur Behandlung, Vorbeugung oder Diagnose von Krankheiten, die zu schwerer Invalidität führen oder lebensbedrohend sind, die in Krisensituationen eingesetzt werden sollen, oder auch für orphan drugs, eine bedingte Zulassung (conditional marketing authorisation) erteilt werden. Dies geht aber nur, wenn das Nutzen-Risiko-Verhältnis des Arzneimittels positiv ist und wahrscheinlich ausreichende klinische Daten nachgereicht werden können. Solche Genehmigungen sind nur ein Jahr gültig, aber verlängerbar. Die Kommission hat hierzu eigens eine Verordnung erlassen ((EG) Nr. 507/2006). Ein Beispiel für eine bedingte Zulassung ist Deltyba (Delamanid) gegen multiresistente Tuberkulose.
Zulassung in Ausnahmefällen
Selbst wenn ein Antragsteller aus objektiven und nachprüfbaren Gründen auch in Zukunft keine vollständigen Daten über die Wirksamkeit und Sicherheit seines Arzneimittels vorlegen kann, ist in Ausnahmefällen unter bestimmten Bedingungen (under exceptional circumstances) eine zentrale Zulassung möglich. Dies kann z.B. bei sehr seltenen Tumorerkrankungen oder kleinen Patientenkollektiven der Fall sein. Die Bedingungen werden jährlich neu beurteilt. Ein Beispiel ist die Zulassung für Aldurazyme (Laronidase) gegen Mukopolysaccharidose I.
Im Verzeichnis der EPARs auf der EMA-Webseite (s. Kasten „Datenbanken“) sind diese Sonderzulassungen erkennbar an den Kürzeln C = conditional, E = exceptional circumstances.
Beschleunigte Zulassung
Humanarzneimittel, die für die öffentliche Gesundheit und besonders als therapeutische Innovation von hohem Interesse sind, können auf Antrag in einem beschleunigten Verfahren (accelerated assessment) bewertet werden. Die Frist für die Beurteilung verkürzt sich dann von 210 auf 150 Tage. Ein Beispiel hierfür ist Soliris (Eculizumab) zur Behandlung der paroxysmalen nächtlichen Hamöglobinurie (PNH).
Compassionate use
Der Off-label-Einsatz eines zugelassenen Arzneimittels für andere Indikationen oder Personengruppen ist gesetzlich nicht geregelt. Anders verhält es sich mit dem „compassionate use“: Nach der Verordnung über das zentrale Zulassungsverfahren können die Mitgliedstaaten nicht zugelassene Humanarzneimittel, die in den Anwendungsbereich des Verfahrens fallen, aus humanen Erwägungen einer Gruppe von Patienten zur Verfügung stellen. Die Patienten müssen allerdings an einer zu Invalidität führenden chronischen, schweren oder lebensbedrohenden Krankheit leiden, und es dürfen keine zufriedenstellenden medikamentösen Alternativen vorhanden sein. Außerdem müssen sich Arzneimittel, die für den „compassionate use“ in Betracht kommen, in einer klinischen Prüfung oder bereits im Zulassungsverfahren befinden. In Umsetzung dieser europäischen Vorgabe wurde der compasionate use mit dem 14. Änderungsgesetz zum Arzneimittelgesetz in das AMG aufgenommen (§ 21 Abs. 2 Nr. 6 AMG). Nähere Verfahrensregelungen sind in der Arzneimittel-Härtefall-Verordnung (gemäß § 80 AMG) niedergelegt. Sie gilt nur für Programme, die auf die Behandlung von Gruppen von Patienten ausgelegt sind. Diese müssen bei der Zulassungsbehörde angezeigt werden. Auf der Webseite des BfArM finden sich ausführliche Erläuterungen zu dem Verfahren sowie eine Liste der genehmigten Compassionate-use-Programme.
Änderungen an zugelassenen Arzneimitteln
Mit der Genehmigung für das Inverkehrbringen werden die Merkmale eines Arzneimittels „modellhaft“ festgelegt. Die Behörde geht davon aus, dass der Zulassungsinhaber das Arzneimittel nur in dieser Form in den Verkehr bringt. Will er Änderungen vorzunehmen, die die Zulassungsunterlagen betreffen, so muss er diese der Zulassungsbehörde anzeigen (§ 29 AMG).
Das Änderungsrecht für Arzneimittel, die in europäischen Verfahren (CP, MRP, DCP) zugelassen wurden, ist in der Verordnung (EG) Nr. 1234/2008 der Europäischen Kommission niedergelegt. Mit dem Zweiten Gesetz zur Änderung arzneimittelrechtlicher und anderer Vorschriften von 2012 wurde der Geltungsbereich der Verordnung auch auf die rein nationalen Zulassungen ausgedehnt (in Deutschland mit bestimmten Ausnahmen). Die Vorschriften sind demnach für europäisch und rein national zugelassene Arzneimittel im Wesentlichen substanziell gleich. Sie unterscheiden sich lediglich in den Modalitäten der Durchführung und der Beteiligung der Behörden. Das System sieht mehrere Änderungstypen vor:
- geringfügige Änderungen (minor variations Typ IA) über ein Mitteilungsverfahren (je nach Art der Änderung entweder unverzüglich oder innerhalb eines Jahres),
- geringfügige Änderungen (minor variations Typ IB) ebenfalls über ein Mitteilungsverfahren, aber mit Zustimmung durch die Behörde. Widerspricht diese innerhalb von 30 Tagen nicht, so gilt die Zustimmung als erteilt (Fiktion).
- größere Änderungen (major variations, Typ II), die ein Genehmigungsverfahren erfordern. Dieses dauert in der Regel 60 Tage.
- Erweiterungen einer Zulassung (line extensions), die nach den Kriterien der Ursprungszulassung behandelt wird (quasi wie eine Neuzulassung). In diesem Fall kann sich der Zulassungsinhaber jedoch soweit möglich auf die vorhandenen Unterlagen zu seinem Arzneimittel beziehen.
- vorläufige Notfallmaßnahmen (urgent safety restrictions) durch den Zulassungsinhaber zur Erhöhung der Arzneimittelsicherheit.
Die Regeln zur Einstufung von Änderungen sind außerordentlich komplex. Die Europäische Kommission hat deshalb eine ausführliche Anleitung für die verschiedenen Kategorien von Änderungen, für die Handhabung der Verfahren sowie zu den jeweils einzureichenden Unterlagen erstellt. Dasselbe gilt für die verwaltungstechnischen Wege zur Unterrichtung der Mitgliedstaaten, in denen das Arzneimittel im Verkehr ist. Auch in der praktischen Umsetzung in einem Pharmaunternehmen ist jede Änderungsanzeige immer wieder eine große Herausforderung. Die gilt insbesondere für multinationale Zulassungen. Die Prozesse müssen engmaschig getaktet und kontrolliert werden. Um eine Verunsicherung und Verwirrung der Patienten zu vermeiden, sollten Präparate, die in mehreren Mitgliedstaaten der EU vermarktet werden, schließlich immer überall auf demselben Status quo gehalten werden.
Literatur
Bei der Verfasserin, auf Anfrage verfügbar.
Autorin
Dr. Helga Blasius, Fachapothekerin für Arzneimittelinformation, Dipl.-Übersetzerin (Jap., Kor.)



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.