













- DAZ.online
- DAZ / AZ
- DAZ 37/2019
- Verdachtsfälle melden

Foto: microstock77 – stock.adobe.com
Pharmakovigilanz
Verdachtsfälle melden
Warum am behördlichen Nebenwirkungsmelde-System kein Weg vorbeiführt
Im Zuge der Zulassung eines Arzneimittels finden umfangreiche Studien zu Wirksamkeit und Sicherheit statt. Tatsächlich erweisen sich aber nur wenige Stoffe während der präklinischen (Testung in vitro und im Tierversuch) und der anschließenden klinischen Phase (Durchführung von klinischen Studien an Probanden/Patienten) als zur Therapie von Patienten geeignet und erhalten eine Zulassung. Gleichwohl ist das Wissen um seine Anwendungsrisiken nicht in vollem Umfang vorhanden.
Grund ist, dass es sich bei klinischen Studien vor Zulassung um interventionelle Studien zur Wirksamkeit handelt: Die Prüfung eines Wirkstoffs erfolgt an einer genau definierten Population, unter vordefinierten Bedingungen und über einen begrenzten, relativ kurzen Zeitraum mit einer eher geringen Patientenzahl. Die Datenlage bei Erteilung der Zulassung hinsichtlich sehr seltener Nebenwirkungen, Risiken in der Langzeitanwendung, Arzneimitteltherapiesicherheit, Wechselwirkungen oder speziellen Risiken für bestimmte Patientengruppen (z. B. Schwangere, Kinder oder ältere Patienten) ist daher noch begrenzt und wird teilweise erst Jahre nach der Zulassung aus der Erfahrung in der breiten Anwendung evident.
Die Meldung von Nebenwirkungsverdachtsfällen ist daher essenziell, mit ihr steht und fällt letztlich die Arzneimittelsicherheit in Europa. Dennoch wird der Meldung von Nebenwirkungen im Alltag von Praxis oder Apotheke, angesichts der sonstigen inhaltlichen und administrativen Pflichten, oft nur wenig Priorität eingeräumt.
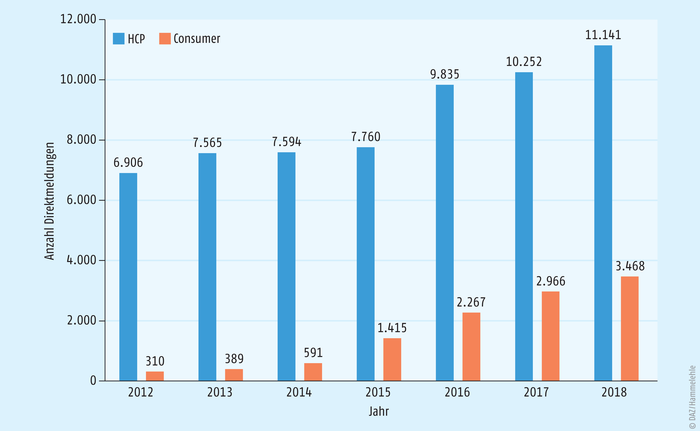
Erfreulicherweise verzeichnet das BfArM in den letzten Jahren eine steigende Meldebereitschaft sowohl bei Angehörigen der Heilberufe (sog. „Healthcare Professionals“, HCP) als auch insbesondere bei Patienten und deren Angehörigen („Consumer“) – s. Abbildung 1.
Dennoch werden nicht alle Nebenwirkungen gemeldet. Die Gründe für dieses sogenannte „Underreporting“ sind vielfältig: So informieren Patienten ihren Arzt nicht über jede Beobachtung, oder der mögliche Zusammenhang zwischen einer Reaktion bzw. deren Symptomen und dem Arzneimittel wird nicht erkannt, z. B. wenn die tatsächlichen Nebenwirkungs-assoziierten Symptome fälschlicherweise der Grunderkrankung zugeordnet werden. Verdachtsfälle von Nebenwirkungen zu Arzneimitteln, die bereits länger auf dem Markt sind, werden weniger häufig gemeldet als solche zu neuen Arzneimitteln.
Abb. 1: Entwicklung der Anzahl von Direktmeldungen durch Angehörige der Heilberufe (Healthcare Professionals; HCP) und Patienten, deren Angehörige oder Vertreter (Consumer) an das BfArM seit 2012. *Anzahl der Bruttomeldungen. Dies entspricht nicht der Anzahl der Nebenwirkungsverdachtsfälle – ein Nebenwirkungsverdachtsfall kann aus mehreren Einzelmeldungen bestehen (z. B. initiale Meldung durch den Patienten, Folgemeldung durch den behandelnden Arzt).
Definition einer Nebenwirkung
Unter einer Nebenwirkung versteht man laut Arzneimittelgesetz (§ 4) jede schädliche und unbeabsichtigte Reaktion auf ein Arzneimittel. In der Humanmedizin ist der Begriff nicht gekoppelt an die Anwendung entsprechend den Vorgaben der Produktinformationen. Auch Reaktionen im Zusammenhang mit einer Arzneimittelanwendung, die nicht den Zulassungsbedingungen entspricht, fallen unter die Definition einer Nebenwirkung, z. B. bei Überdosierung, Medikationsfehlern oder dem „Off-Label-Use“, d. h. der Anwendung außerhalb der Zulassungsbedingungen im Sinne der ärztlichen Therapiefreiheit [1].
Meldepflichten in Deutschland
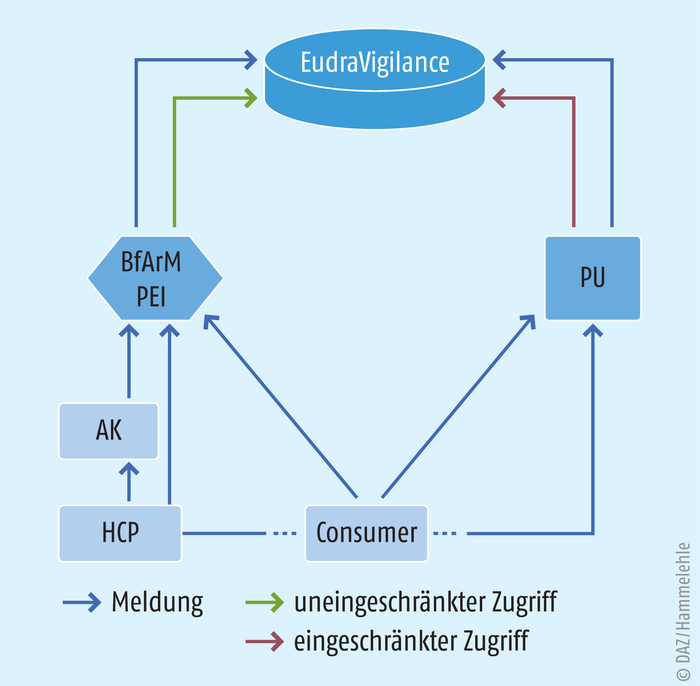
In Deutschland gibt es für Ärzte und Apotheker laut Arzneimittelgesetz keine Verpflichtung, Nebenwirkungen zu melden. Die Pflicht zur Meldung ergibt sich aber aus der jeweiligen Berufsordnung [2]. Adressat ist dabei die Arzneimittelkommission der jeweiligen Kammer. Diese wiederum geben die Meldungen an die jeweils zuständige Bundesoberbehörde weiter, unterstützen also mit ihren Meldesystemen unmittelbar das behördliche Pharmakovigilanzsystem (s. Abb. 2).
Abb. 2: Übersicht über das deutsche Meldesystem von Nebenwirkungsverdachtsfällen im europäischen Austausch HCP = Healthcare Professionals (Angehörige der Heilberufe); Consumer = Patienten / deren Angehörige oder deren Vertreter; PU = Pharmazeutischer Unternehmer; AK = Arzneimittelkommissionen der Heilberufskammern
Oft stehen Ärzte und Apotheker vor der Frage, ob sie denn jeden Nebenwirkungsverdachtsfall melden sollen und müssen. Die Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) und die Arzneimittelkommission der Deutschen Apotheker (AMK) beispielsweise haben einen entsprechenden Leitfaden publiziert [3, 4].
Von besonderem wissenschaftlichem Interesse sind sämtliche schwerwiegende, unbekannte (also in der Produktinformation nach Art, Ausmaß oder Häufigkeit nicht beschriebene) Reaktionen auf ein Arzneimittel.
Aber auch die Meldung bekannter, nicht schwerwiegender Nebenwirkungen kann wichtig sein, wenn z. B. die Therapie-Compliance des Patienten durch diese Nebenwirkungen beeinträchtigt wird.
Bei biologischen Arzneimitteln (Biologika [6]) ist die Nennung der Chargennummer ergänzend zu beachten. Um Chargen-assoziierte Nebenwirkungen mit entsprechendem Gefahrenpotenzial, z. B. Übertragung von Infektionen, gezielt nachverfolgen zu können, besteht der gesetzliche Auftrag für pharmazeutische Unternehmen und Behörden zu versuchen, diese in Erfahrung zu bringen.
Spontan berichtete Nebenwirkungen (d. h. die Meldung stammt nicht aus systematisierten Untersuchungen, in denen u. a. Nebenwirkungen erfasst werden) müssen keine explizite Kausalitätsbeurteilung durch den Meldenden enthalten. Hier ist der Verdacht auf einen kausalen Zusammenhang zwischen dem Arzneimittel und der aufgetretenen Nebenwirkung bereits durch die Meldung selbst impliziert, da sie andernfalls wohl nicht erfolgt wäre. Dennoch ist die medizinische Beurteilung eines Falles durch den behandelnden Arzt, der den betroffenen Patienten und seine Krankengeschichte kennt, eine für die Behörden wertvolle und im Einzelfall nicht ersetzbare Information.
Von dieser generellen Betrachtungsweise der Kausalität bei Spontanberichten wird nur dann abgewichen, wenn der Meldende einen solchen Zusammenhang ausdrücklich verneint. Der Begriff des „Verdachts“ bedeutet gleichzeitig, dass ein kausaler Zusammenhang nicht bewiesen sein muss, bevor eine Meldung erfolgt. Ein lückenloser Beweis wird nur in den seltensten Fällen geführt werden können, wenn überhaupt.
Patienten und deren Angehörige können den Verdacht einer Arzneimittelnebenwirkung ebenfalls melden. Eine begleitende ärztliche Beurteilung und Ergänzung mit weiteren Daten, die dem Patienten evtl. nicht vorliegen, ist wünschenswert, aber keine zwingende Voraussetzung. In die Beurteilung von Arzneimittelrisiken geht jede Meldung in gleicher Weise ein, unabhängig von der Quelle und unabhängig von der medizinischen Qualifikation der meldenden Person.
Meldungen sollten unter Angabe der Meldequelle und pseudonymisiert bezogen auf den Patienten erfolgen, sofern der Patient nicht selbst die Meldequelle darstellt. In vielen Fällen sind medizinische Rückfragen notwendig, um einen Bericht besser beurteilen zu können. Die Pseudonymisierung ist aus datenschutzrechtlichen Gründen erforderlich, erlaubt aber auch, Informationen aus unterschiedlichen Quellen, aber zum selben Fall, zusammenzuführen, was ohne diese Angaben nicht verlässlich möglich ist.
AkdÄ und AMK warnen vor „Nebenwirkungen.de“
Die Aktivitäten des privaten Anbieters Medikura mit seinem Portal Nebenwirkungen.de, der nicht nur Patienten, sondern inzwischen auch gezielt Apotheker und Ärzte zur Meldung von Nebenwirkungen auffordert, haben die Arzneimittelkommission der deutschen Ärzte (AkdÄ) und die der Deutschen Apotheker (AMK) auf den Plan gerufen. So stößt bei der AkdÄ die Verarbeitung und Vermittlung von sensiblen medizinischen Daten von einzelnen Patienten im Zusammenhang mit der Arzneimittelsicherheit durch ein gewinnorientiertes Unternehmen der Datenverarbeitung auf vehemente Ablehnung. Es sei nicht ersichtlich, welchen Nutzen Medikura zum bestehenden Meldesystem beisteuert. „Die Arzneimittelsicherheit ist ein Anliegen der öffentlichen Gesundheit. Sie sollte in den Händen öffentlicher und nicht gewinnorientierter Organisationen verbleiben“, so die AkdÄ.
Auch die AMK hat sich inzwischen entsprechend positioniert und macht noch einmal ihre Rolle in dem behördlichen System klar: „Als Heilberufler sind Apothekerinnen und Apotheker aufgrund ihrer Berufsordnung verpflichtet, bei der Ermittlung, Erkennung, Erfassung und Weitergabe sowie an der Vorbeugung von Arzneimittelrisiken aktiv mitzuwirken und Arzneimittelrisiken an die AMK zu melden. So ist die Apothekerschaft in das nationale Pharmakovigilanzsystem auch formal eingebunden.“
Unabhängig davon begrüßt die AMK die Möglichkeit für Patienten und Angehörige, ihre Nebenwirkungsverdachtsfälle direkt bei den Bundesoberbehörden melden zu können (www.nebenwirkungen.pei.de). Das Start-up-Unternehmen Medikura mit seinem Portal Nebenwirkungen.de ist der AMK jedoch ein Dorn im Auge. Kritisiert wird, dass sich das Unternehmen zunehmend öffentlichkeitswirksam als „digitale und innovative Infrastruktur für den Austausch von Arzneimittelwirkungen“ in Konkurrenz zu den etablierten Meldestrukturen bewirbt und insbesondere Nebenwirkungsmeldungen von Patienten erhalten möchte. Die AMK ist davon überzeugt, dass das Geschäftsmodell von Medikura darauf abzielt, die über die Meldeplattform gesammelten Daten gewinnbringend an interessierte Stellen zu verkaufen. Dieses Vorgehen, also die Erfassung und Weiterleitung hochsensibler gesundheitsbezogener Daten durch gewinnorientierte privatwirtschaftliche Unternehmen, lehnt die AMK strikt ab.
Zudem kann die AMK nicht erkennen, wie so das Problem des „Underreportings“, also der unzureichenden Bereitschaft zu melden, behoben werden kann. Im Gegenteil: Unterschiedliche Meldesysteme würden die Gefahr von Doppelmeldungen erhöhen und zu Duplikaten im europäischen Pharmakovigilanzsystem führen.
Die AMK weist in ihrer Stellungnahme noch einmal ausdrücklich darauf hin, dass die Nutzung von Nebenwirkungen.de Apothekerinnen und Apotheker nicht von ihrer Pflicht entbinden, ihnen bekannt gewordene Risiken unverzüglich an die zuständige Überwachungsbehörde bzw. die AMK zu melden.
daz
Quelle:
Position der AkdÄ zum Online-Portal „Nebenwirkungen.de“ des Unternehmens Medikura. 1. August 2019 https://www.akdae.de/Stellungnahmen/Weitere/20190802.pdf
Meldungen von Nebenwirkungen über Medikura. Position der Arzneimittelkommission der Deutschen Apotheker (AMK), vom 27. August 2019
Die Pflichten der Unternehmer
Angehörige der Heilberufe und Patienten können Nebenwirkungsmeldungen auch gegenüber den Zulassungsinhabern vornehmen. Diese sind nach dem Arzneimittelgesetz verpflichtet, jeden ihnen bekannt gewordenen Nebenwirkungsfall direkt an die Europäische Nebenwirkungsdatenbank (EudraVigilance) bei der Europäischen Arzneimittel-Agentur (EMA) zu melden.
Die in EudraVigilance gespeicherten Daten sind der Öffentlichkeit in aufgearbeiteter und datenschutzrechtlich zulässiger Form unter http://www.adrreports.eu zugänglich.
Melden an das BfArM und das PEI
Nebenwirkungsmeldungen können in jedweder Form, sollten aber insbesondere elektronisch an das BfArM herangetragen werden.
Die bequemste und aus Sicht der Behörden zu bevorzugende Möglichkeit, Nebenwirkungen zu melden, ist die Meldung über das Onlineangebot der beiden in Deutschland zuständigen Oberbehörden BfArM und PEI:
- Für Angehörige der Heilberufe ist eine Nebenwirkungsmeldung bereits seit über 10 Jahren online über https://humanweb.pei.de möglich.
- Patienten steht seit 2012 ein Online-Meldeportal zur Verfügung, welches im September 2018 eine umfangreiche Neugestaltung erfahren hat [5] und den Nutzer anwenderfreundlich und intuitiv durch den Prozess führt (https://nebenwirkungen.pei.de).
Diese beiden Portale sind die einzigen in Deutschland von den zuständigen Behörden entwickelten und geprüften Online-Angebote zur Erfassung von Nebenwirkungen. Auf Wunsch wird dem Meldewilligen aber auch ein Papier-Formular zur Verfügung gestellt, das handschriftlich ausgefüllt und an das BfArM zurückgesendet wird. Die Möglichkeit der Meldung an das BfArM ist damit unabhängig von den vorhandenen technischen Gegebenheiten oder der Bereitschaft zur Nutzung digitaler Medien.
Private Anbieter ohne behördliche Kontrolle
Im Internet finden sich vereinzelt Seiten privater Anbieter, die Patienten dazu auffordern, Nebenwirkungsmeldungen auf diesen Seiten abzugeben. Es wird damit geworben, dass diese Meldungen den pharmazeutischen Unternehmen und behandelnden Ärzten unmittelbar weitergeleitet und somit den direkten Informationsaustausch gewährleisten würden.
Diese Angebote unterliegen keiner Kontrolle durch die mit dem Schutz der Arzneimittelsicherheit beauftragten Bundesoberbehörden BfArM und PEI, sodass durch die Aufsichtsbehörden nicht nachvollzogen werden kann, was mit den dort abgegebenen Informationen geschieht. Eine Rechtsgrundlage zur Inspektion der Betreiber solcher Plattformen ist nicht gegeben. Auch kann das BfArM, das entsprechend seines gesetzlichen Auftrags jeden Verdachtsfall einer Nebenwirkung, gleich welcher Form und welchen Inhalts, aufnehmen und verarbeiten muss, diesen Anbietern keinerlei Qualitätsvorgaben zu ihren Meldungen machen.
Verarbeitung von Verdachtsfällen im BfArM
Jeder beim BfArM eingegangene Verdachtsfall einer Nebenwirkung wird in einer dafür entwickelten Nebenwirkungsdatenbank erfasst. Die Erfassung erfolgt größtenteils manuell nach internationalen Standards. Dabei wird der Informationsgehalt einer frei formulierten Nebenwirkungsmeldung strukturiert (z. B. nach den Angaben zu Arzneimitteln, Nebenwirkungen, Begleiterkrankung) und in standardisierten Datenfeldern hinterlegt. Mittels dieser technischen Aufbereitung ist es möglich, dass die abgegebenen Einzelfallmeldungen überall in Europa (aber auch, ggf. mit Adaptionen, in anderen Regionen der Welt) gelesen, verstanden und analysiert werden können.
Bei Meldung von Nebenwirkungen an das BfArM können alle Beteiligten darauf vertrauen, dass ihre Meldungen den weltweit anerkannten Standards entsprechen und die enthaltenen Informationen für eine spätere medizinische Beurteilung wissenschaftlich belastbar aufbereitet werden. Dies schließt ein, dass der Meldungsinhalt sowohl vertraulich entsprechend der geltenden datenschutzrechtlichen Bestimmungen als auch mit der nötigen Expertise behandelt wird. Insgesamt ist somit sichergestellt, dass jede einzelne Meldung unter Beachtung aller relevanten Rahmenbedingungen der fortlaufenden Bewertung der Anwendungssicherheit von Arzneimitteln auf europäischer Ebene zugeführt wird und damit einen Beitrag zur Verbesserung der Arzneimittelsicherheit darstellt.
Privatanbieter, die basierend auf einem kommerziell ausgelegten Geschäftsmodell für die Erfassung von Nebenwirkungen werben, einer behördlichen Kontrolle aber nicht unterworfen sind, können diese Sicherheit aus Sicht des BfArM nicht geben.
Beurteilung von Nebenwirkungsverdachtsfällen
Der Großteil der sich heute auf dem Markt befindlichen Arzneimittel ist über europäische Verfahren, d. h. länderübergreifend, zugelassen worden. Entsprechend ist auch die Überwachung der Arzneimittelsicherheit (Pharmakovigilanz) europäisch ausgerichtet. Eine rein national gestützte Arzneimittelsicherheit würde nur einen kleinen Teil der in Europa exponierten Patienten berücksichtigen, eine enge europäische Zusammenarbeit in der Pharmakovigilanz ist daher zwingend notwendig.
Daher wird jeder dem BfArM gemeldete Verdachtsfall einer Nebenwirkung an EudraVigilance unter Beachtung der datenschutzrechtlichen Bestimmungen weitergeleitet.
Nur selten ist es möglich, aufgrund eines einzelnen Verdachtsfalles eine umfassende Beurteilung des Anwendungsrisikos durchzuführen und sofortige Maßnahmen einzuleiten. Daher werden die in EudraVigilance gespeicherten Nebenwirkungsmeldungen durch Experten der EMA und der nationalen Behörden fortlaufend analysiert. Daraus können sich Hinweise („Signale“) für bislang unbekannte Nebenwirkungen im Zusammenhang mit der Anwendung eines bestimmten Arzneimittels bzw. Wirkstoffes oder einer Wirkstoffkombination ergeben. Die Bewertung dieses Signals wird durch die Vertreter von EMA und nationalen Behörden, einschließlich des BfArM, unter Einbindung des europäischen Expertengremiums, dem Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz („Pharmacovigilance Risk Assessment Committee“, PRAC) durchgeführt. Pharmazeutische Unternehmer sind dabei aufgefordert, ggf. weitere Informationen zur Verfügung zu stellen. Die beim PRAC diskutierten Signale werden auf der Webseite der EMA publiziert, ebenso die Ergebnisse der Bewertung durch den PRAC in Form von Empfehlungen.
Die Überwachung der Anwendungssicherheit durch die Bewertung von Signalen wird ergänzt durch die Bewertung der vom Zulassungsinhaber vorzulegenden periodischen Sicherheitsberichte (engl. „Periodic Safety Update Report“, PSUR) sowie die Durchführung von Risikobewertungsverfahren, wenn das Nutzen-Risiko-Verhältnis von Arzneimitteln bzw. Wirkstoffen/Wirkstoffkombinationen infrage steht. Aus diesen Überprüfungen können Maßnahmen zur Minimierung des Anwendungsrisikos abgeleitet werden, die auf europäischer Ebene festgelegt werden und die dann in den Mitgliedsländern verbindlich umzusetzen sind. Das Spektrum möglicher Maßnahmen reicht von Änderungen der informativen Texte in Fach- und Gebrauchsinformation, der Anordnung zur Erstellung weiterer Informationsmaterialien für Ärzte und/oder Patienten (sog. Schulungsmaterial), die Anordnung zur Durchführung weiterer Studien durch die Zulassungsinhaber, die Information der Fachkreise über neue Anwendungsrisiken mit Auswirkungen auf das Verordnungsverhalten mittels „Rote-Hand-Briefe“ bis hin zum Widerruf einer Zulassung, wenn das Nutzen-Risiko-Verhältnis negativ ist, d. h. das Anwendungsrisiko im Verhältnis zum Nutzen eines Arzneimittels als nicht mehr vertretbar erscheint. |
Literatur
[1] Lütkehermölle W, Paeschke N: Einführung in die Grundlagen der Pharmakovigilanz (Teil I) – Verdachtsfälle von unerwünschten Arzneimittelwirkungen. Bulletin zur Arzneimittelsicherheit von BfArM und PEI, 1/2010:14-17
[2] Stammschulte T, Pachl H, Gundert-Remy U, Lütkehermölle W, Paeschke N, Volz-Zang C, Keller-Stanislawski B: Einführung in die Grundlagen der Pharmakovigilanz (Teil II): Spontanmeldesystem zur Erfassung von Verdachtsfällen unerwünschter Arzneimittelwirkungen (UAW). Bulletin zur Arzneimittelsicherheit von BfArM und PEI, 4/2010:18-26
[3] https://www.akdae.de/Arzneimitteltherapie/LF/PDF/Nebenwirkungen_melden.pdf; abgerufen am 10.07.2019
[4] Zagermann-Muncke P, Frölich S, Schulz M: Pharmakovigilanz: Unerwünschte Wirkungen an die AMK melden. Pharmazeutische Zeitung 10/2010:16-23
[5] Ruhaltinger D, Stoll H, Mentzer D, Alesik E, Huber M: „Online-Meldung von Arzneimittelnebenwirkungen“ Bulletin zur Arzneimittelsicherheit von BfArM und PEI, 4/2018:20-23
[6] Definition Biologika: Gemäß Anhang I, Teil I, Art. 3.2.1.1 der Richtlinie 2001/83/EG ist ein biologisches Arzneimittel ein Arzneimittel, dessen Wirkstoff eine biologische Substanz ist, d. h. eine Substanz, die von einer biologischen Quelle gewonnen wird und zu deren Charakterisierung und Qualitätsbestimmung physikalische, chemische und biologische Verfahren sowie eine Beurteilung des Produktionsprozesses sowie dessen Kontrolle eingesetzt werden. Diese umfassen immunologische Arzneimittel, aus menschlichem Blut und Plasma gewonnene Arzneimittel entsprechend den Definitionen in Art. 1 Abs. 4 und 10, Arzneimittel, die unter Teil A des Anhangs der Verordnung (EWG) Nr. 2309/93 fallen und die in Teil IV dieses Anhangs definierten Arzneimittel für neuartige Therapien.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.