














- DAZ.online
- DAZ / AZ
- DAZ 43/2018
- The product is the ...

Foto: Ivan Traimak - stock.adobe.com
Schwerpunkt Biosimilars
The product is the process
Was der Herstellungsprozess der Biosimilars für die Zulassung bedeutet
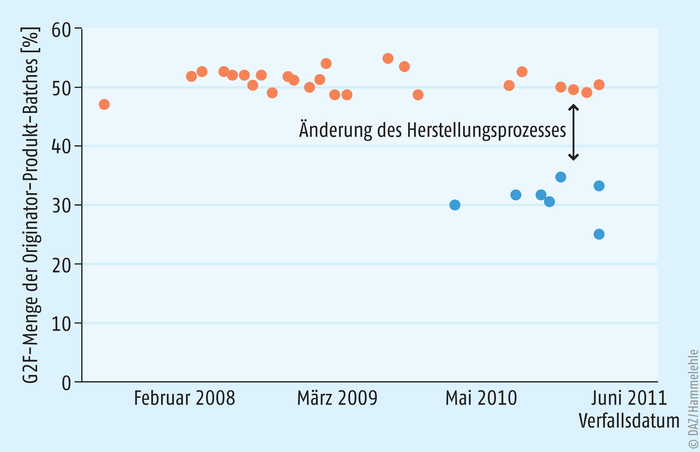
Biosimilars sind keine Generika. Denn während Generika monomolekular vorliegen und daher tatsächlich kopiert werden können, ist es ein inhärentes Merkmal aller biologischer Produkte, dass sie immer in Form komplexer Mischungen von Molekülen vorliegen, deren Einzelmoleküle sich von dem intakten Biomolekül in Nuancen unterscheiden. Diese Mikroheterogenität resultiert aus Varianten, die als Artefakte zu bezeichnen sind und die für alle Biopharmazeutika immer in mehr oder weniger engen, klar spezifizierten Konzentrationskorridoren vorhanden sind (Abb. 1).
Das Prinzip der inhärenten Mikroheterogenität gilt für Biosimilars ebenso wie für die als Kopiervorlage genutzten Referenzarzneimittel. Die Konsequenz ist, dass ein Biosimilar ähnlich, aber niemals identisch mit dem Referenzarzneimittel sein kann. Allerdings sind die strukturellen Abweichungen des Biosimilars von seinem korrespondierenden Referenzarzneimittel keinesfalls beliebig. Sie dürfen nicht größer sein als die Abweichungen innerhalb verschiedener Chargen des Referenzproduktes. Tatsächlich sind auch verschiedene Chargen der als Referenz gewählten Innovatorprodukte immer „nur“ ähnlich und nie zueinander identisch.
Biosimilar und Referenzarzneimittel besitzen zwingend die identische Aminosäuresequenz. Bei diesem Kriterium sind keine Unterschiede zwischen dem „Original“ und der „Kopie“ zulässig. Allerdings ist es möglich, dass einzelne Aminosäuren durch Umwelteinflüsse (pH-Wert, Temperatur, Druck) modifiziert sind (z. B. oxidierte Varianten von Methionin, Cystein, Tryptophan, Tyrosin und Histidin; deamidierte Varianten von Asparagin bzw. Glutamin; hydrolysierte Disulfidbrücken). Ferner können sich Details des Glykosylierungsmusters sowohl zwischen „Original“ und „Kopie“ als auch zwischen unterschiedlichen Chargen von „Original“ und „Kopie“ unterscheiden. Diese Variationen sind Teil der generellen Mikroheterogenität von Biopharmazeutika und somit auch von Biosimilar und Referenzarzneimittel.
Der Grundsatz „the product is the process“ gilt streng für die Herstellung aller Biopharmazeutika und damit auch für Biosimilars. Allerdings muss der Prozess nicht mit dem Prozess identisch sein, mit dem die Wirkstoffchargen hergestellt wurden, die in den Zulassungsstudien eingesetzt wurden. Prozesse können heute geändert oder sogar neu aufgesetzt werden, vorausgesetzt, die Änderungen wurden der EMA gemeldet und von ihr genehmigt.
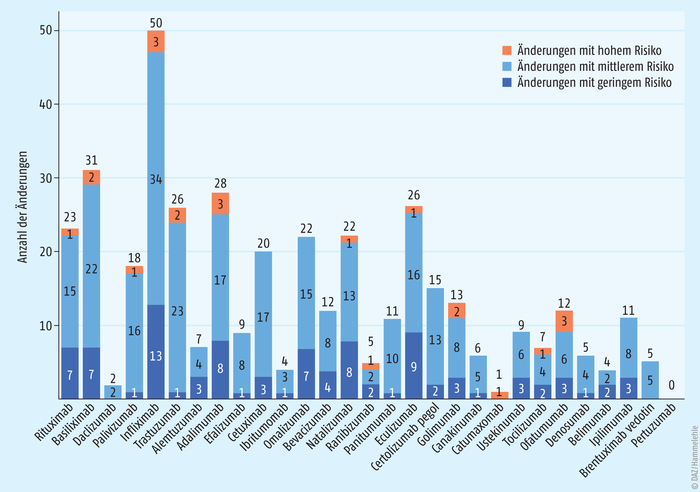
Dies ist eindrucksvoll in einer jüngeren Studie dokumentiert (Abb. 2). Hier wurde die Anzahl der Änderungen der Herstellungsprozesse von 29 monoklonalen Antikörpern ermittelt. 404 Prozessmodifikationen wurden in den öffentlich zugänglichen Zulassungsinformationen (EPAR, European public assessment reports) der EMA gefunden – davon allein 50 für die Herstellung des Originator-Arzneimittels Remicade® (INN: Infliximab). 32 dieser Änderungen waren gravierende Eingriffe wie ein Wechsel der Zelllinie für die Wirkstoffproduktion. Im Durchschnitt erfolgten 1,8 Änderungen des Produktionsprozesses pro Jahr und Biopharmazeutikum [1].
Die Genehmigung einer Prozessänderung für ein Originator-Biopharmazeutikum wird durch die EMA auf der Basis von Daten erteilt, die der pharmazeutische Hersteller erheben und der Behörde übermitteln muss. Diese Daten stammen aus Überbrückungsstudien (bridging studies). Dazu werden Wirkstoff-Chargen aus dem Ursprungsprozess mit Wirkstoff-Chargen aus dem modifizierten Prozess verglichen. Das Verfahren wird als comparability exercise bezeichnet. Die wichtigsten Methoden in diesen Verfahren sind bioanalytischer Natur, da durch sie potenzielle Unterschiede mit der höchsten Sensitivität nachgewiesen werden können. Deshalb sind diese Methoden von besonderer Bedeutung im Rahmen einer comparability exercise. Ergänzt werden diese Analysen durch präklinische Tests und – sofern aus regulatorischer Sicht erforderlich – auch durch klinische Studien. Nach ganz analogen Prinzipien wie bei der Durchführung von Überbrückungsstudien werden im Rahmen einer comparability exercise auch Referenzarzneimittel und Biosimilar miteinander verglichen und hinsichtlich ihrer Ähnlichkeit bewertet.
Wird eine Prozessänderung genehmigt, betrifft dies alle Indikationen, für die der Wirkstoff zugelassen ist. Dies gilt auch dann, wenn eine für notwendig erachtete klinische Studie im Rahmen der comparability exercise nur in einer (sensitiven) Indikation durchgeführt wurde. Diesem Vorgehen liegt das Prinzip der Extrapolation der Indikation zugrunde, das bei Biopharmazeutika seit Jahren angewandt wird und sich seit Jahren bewährt hat, selbst dann, wenn über größere Prozessänderungen entschieden werden musste. Beispielsweise wurde die subkutane Applikationsform des monoklonalen HER2-Antikörpers Trastuzumab (Herceptin®) auf Basis von Daten von Patientinnen mit metastasierter Erkrankung auch für die neoadjuvante Therapie zugelassen.
Sein immunologisches Potenzial ist ein wichtiger Aspekt bei der Beurteilung eines Biopharmazeutikums. Nahezu alle Biopharmazeutika induzieren beim Patienten die Bildung von Antikörpern, die gegen das Therapeutikum gerichtet sind (Anti-Drug-Antikörper, ADA). Diese können das therapeutische Verhalten des Biopharmazeutikums verändern, besonders dann, wenn es sich bei den Anti-Drug-Antikörpern um neutralisierende Antikörper handelt. In seltenen Fällen können Anti-Drug-Antikörper auch die Sicherheit eines Arzneimittels beeinträchtigen, indem sie vermehrt Hypersensitivitätsreaktionen, Anaphylaxie oder Immunkomplexkrankheiten hervorrufen [2].
Für alle Biopharmazeutika wird das immunogene Potenzial bei jeder Indikation durch klinische Studien ermittelt. Zudem sind Antikörpertests stets Bestandteil klinischer Prüfungen von Biopharmazeutika. Wahrscheinlich wegen dieser hohen Sensibilität gegenüber dem potenziellen immunologischen Problem gibt es bisher kein Beispiel dafür, dass durch Prozessänderungen, die nicht zur Neudefinition der ursprünglichen Mikroheterogenitätskorridore geführt haben, eine erhöhte Immunogenität resultierte, die zu schwerwiegenden Nebenwirkungen geführt hätte. Dies gilt gleichermaßen für Biosimilars.
Zulassungssystematik für Biosimilars
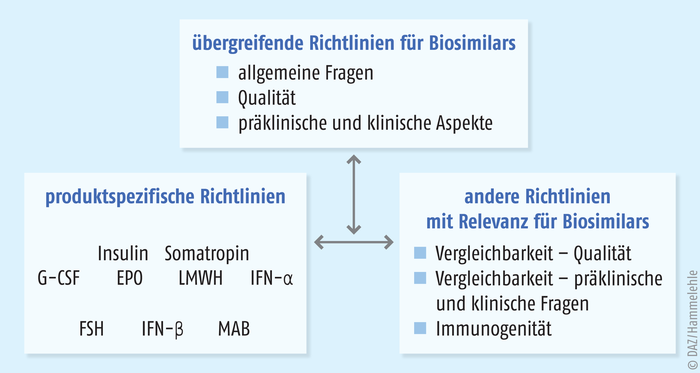
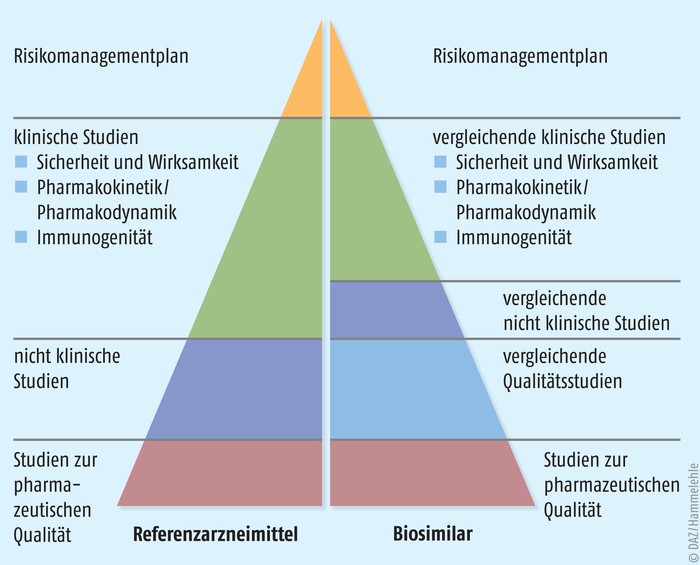
Alle gentechnisch hergestellten Arzneimittel müssen in einem zentralisierten Verfahren über die Europäische Arzneimittelbehörde (European Medicines Agency, EMA) zugelassen werden. Damit müssen auch Biosimilars bei dieser Behörde ein spezielles Zulassungsverfahren durchlaufen, in dem die Qualität, die Wirksamkeit, die Sicherheit und die Verträglichkeit der Präparate nach festen Regeln überprüft werden. Die Basis für dieses seit 2005 implementierte Verfahren bilden die zu diesem Zweck angepassten Direktiven 2001/83/EC und 2003/63/EC. Die Zulassungsanforderungen für Biosimilars sind derzeit in 15 Leitlinien beschrieben – drei übergeordnete Leitlinien, acht Produktspezifische Leitlinien und vier andere Leitlinien [3]. Durch dieses Zulassungsverfahren (Abb. 3) unterscheiden sich Biosimilars auch grundlegend von anderen, im globalen Markt verfügbaren Nachahmerprodukten eines Biopharmazeutikums. Ohne eine durch die EMA erteilte Zulassung sind diese Biosimilars in Europa nicht verkehrsfähig und zeigen zum Teil gefährliche Qualitätsmängel. Auch passiert nicht jeder zur Zulassung bei der EMA eingereichte Biosimilar-Kandidat die anspruchsvollen Anforderungen des zentralisierten Verfahrens zum Nachweis einer Gleichwertigkeit mit dem Referenzarzneimittel. Dies belegt die hohen Ansprüche, die an diese Arzneimittel gestellt werden, und es unterstreicht die Gleichwertigkeit von Biosimilars mit den entsprechenden Referenzprodukten nicht nur hinsichtlich ihrer Wirksamkeit, sondern auch hinsichtlich ihrer Qualität und Unbedenklichkeit. Gleichzeitig zeigt die Tatsache, dass nicht jeder Biosimilar-Kandidat die anspruchsvollen Anforderungen des zentralisierten Verfahrens erfüllt, wie wichtig es ist, dass nur in der EU zugelassene Biosimilars therapeutisch eingesetzt werden dürfen.
Der Herstellungsprozess
Der Herstellungsprozess kann in vier aufeinander folgende Schritte unterteilt werden. Das Ziel ist, nicht etwa ein neues, in seinen klinischen Eigenschaften noch gänzlich unbekanntes Molekül zu entwickeln, sondern ein Molekül, das über Jahre klinisch erprobt ist (Referenzarzneimittel), bestmöglich zu kopieren, sodass alle verfügbaren Daten zum Referenzprodukt auch für die Kopie (Biosimilar) genutzt werden können.
Im ersten Schritt muss der Hersteller eines Biosimilars die Referenzarzneimittel bis ins kleinste Detail analysieren, um konkrete Vorstellungen davon zu bekommen, was kopiert werden muss. Hierzu erwirbt der Hersteller des Biosimilars auf den relevanten Märkten das Referenzarzneimittel in großer Zahl, wobei möglichst viele unterschiedliche Chargen repräsentiert sein sollten. Diese werden mit modernsten bioanalytischen Methoden hinsichtlich hunderter verschiedener Strukturcharakteristika qualitativ wie quantitativ analysiert. Zu diesen Charakteristika gehören auch die bereits erwähnten Strukturmodifikationen wie oxidierte oder deamidierte Varianten verschiedener Aminosäuren, modifizierte N- und C-terminale Enden der Proteinketten, Details der Glykosylierungscharakteristika, der Anteil an denaturiertem Protein, Protein-Aggregate und viele mehr. Dies ist erforderlich, da auch diese Strukturvarianten kopiert werden müssen. Natürlich darf das Biosimilar nicht schlechter sein als das Referenzprodukt. Es darf aber auch nicht besser sein.
Im zweiten Schritt muss der Biosimilar-Hersteller auf Basis der von ihm erhobenen Analysedaten für das Referenzarzneimittel Variationskorridore festlegen, in denen die Mikroheterogenität toleriert ist. Die Ober- und Untergrenzen dieser Spezifikationskorridore dürfen nicht die entsprechenden Grenzen des Referenzproduktes über- oder unterschreiten. Natürlich kennt der Biosimilar-Hersteller die Grenzen der Spezifikationskorridore des Referenzarzneimittels nicht, sodass er gezwungen ist, sich bei diesem wichtigen Schritt auf seine eigenen Analysen des Referenzwirkstoffs zu verlassen.
Andererseits kennt natürlich die Zulassungsbehörde alle Details zum Referenzarzneimittel und damit auch die Spezifikationen der Mikroheterogenitätskorridore. So ist es nicht ungewöhnlich, dass die Spielräume für die Mikroheterogenität bei Biosimilars oft enger ausfallen, als es theoretisch möglich wäre.
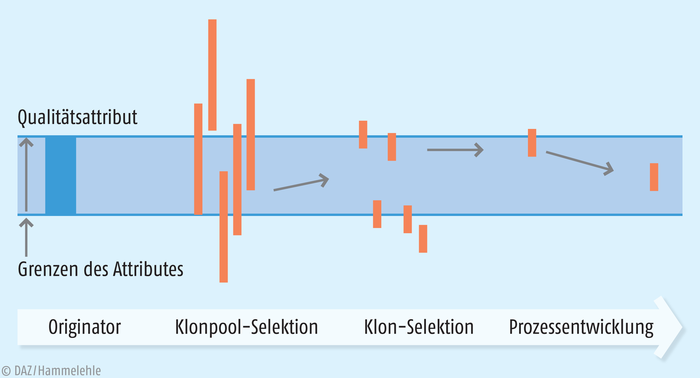
Im dritten Schritt muss nun das Biosimilar mithilfe der komplexen Methoden der Biotechnologie hergestellt werden. Dazu werden zunächst verschiedene Produktionsklone dahingehend getestet, in wie weit sie Produkte liefern, die möglichst gut dem Referenzprodukt ähneln. Durch aufwendige Zyklen von Prozessvariation und Bioanalytik der jeweils resultierenden Produkte nähert man sich allmählich den Bedingungen, die sicherstellen, dass das Biomolekül dem Referenzarzneimittel in allen Vorgaben ähnelt (Abb. 4).
Dieser Prozess wird dann in jedem denkbaren Detail spezifiziert und bildet nach dem Prinzip „the product is the process“ die Basis für die Herstellung aller Chargen des Biosimilars.
Im vierten Schritt erfolgt dann die umfassende Überprüfung des Biosimilars – der Prozess, der als comparability exercise beschrieben wird. Ein entscheidendes Charakteristikum dieses Teils besteht darin, dass alle Tests head-to-head zwischen Biosimilar und Referenzarzneimittel durchgeführt werden. Dies betrifft auch die erforderliche klinische Studie am Ende dieses Vergleichsprozesses.
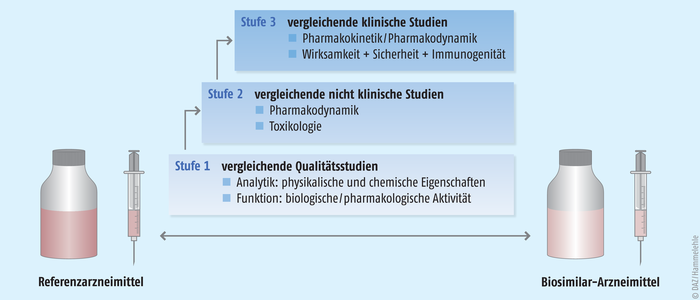
Auch diese Verfahren sind sequenziell ausgelegt (Abb. 5).
Zunächstwerden die Vergleichbarkeit der Qualität bzw. die physikalisch-chemische und die biologische Vergleichbarkeit demonstriert. Hier wird die strukturelle Übereinstimmung von Biosimilar und Referenzarzneimittel in allen relevanten Details mit einem riesigen Spektrum an analytischen Methoden belegt. Mögliche Abweichungen von den Daten des Referenzproduktes müssen plausibel erklärt werden. Ferner wird die Reinheit des Biosimilars überprüft. Das Produkt wird nur freigegeben, wenn im Vorfeld definierte Spezifikationskriterien erfüllt werden.
Anschließendwerden Biosimilar und Referenzarzneimittel im präklinischen Setting miteinander verglichen. In aller Regel kann hier auf ein verkürztes Verfahren in Form von In-vitro-Untersuchungen zugegriffen werden. Diese Untersuchungen sind in produktspezifischen Richtlinien durch die EMA vorgegeben. Die Pharmakokinetik- und die Pharmakodynamik-Parameter sowie deren vordefinierter Grad der Ähnlichkeit mit dem Referenzprodukt müssen begründet und getroffen werden.
Schließlich wird dann die klinische Vergleichbarkeit belegt. Diese Studien haben mehr den Charakter von Sicherheitsstudien als von Wirksamkeitsstudien. Wenn im ersten Schritt der umfassenden Überprüfung belegt ist, dass Biosimilar und Referenzarzneimittel ausreichend ähnlich sind, dann ist auch damit zu rechnen, dass sie klinisch äquivalent wirken. Zwingend sind diese klinischen Studien jedoch gefordert, um die Verträglichkeit der Biosimilars zu belegen. Denn die Herstellungsprozesse von Biosimilar und Referenzprodukt sind zwangsläufig unterschiedlich, sodass nicht ausgeschlossen werden kann, dass analytisch nicht oder nur schwer fassbare Komponenten eine klinische Auffälligkeit provozieren. In klinischen Phase-I-Studien liegt der Fokus zunächst auf der Toxikologie, der Pharmakokinetik und der Pharmakodynamik. Das heißt, der Wirkstoff wird auf seine Reinheit und Unbedenklichkeit hin geprüft, seine Charakteristika im Körper nachvollzogen (Aufnahme, Verteilung im Körper, biochemischer Auf- und Umbau sowie Ausscheidung) und ein Wirkprofil erstellt. Daran schließen sich Studien zur Wirksamkeit und Sicherheit im Sinne von Schwere und Häufigkeit verschiedener Nebenwirkungen bei einer oder mehreren repräsentativen Indikationen an, um ein vergleichbares Wirksamkeits- und Sicherheitsprofil zu demonstrieren. Hierzu zählt auch ein vergleichbares Immunogenitätsprofil von Biosimilar und Referenzarzneimittel. Schwerpunkte und Anforderungen an diese Phase-III-Studien sind je nach Biosimilar-Klasse verschieden. Entsprechend der Unterschiedlichkeit und Komplexität von biologischen Pharmazeutika legt die EMA die Anforderungen jedoch individuell und an den Leitlinien orientiert mit den Herstellern fest (Abb. 6). |
Literatur
[1] Vezer B, Buzas Z, Sebeszta M, Zrubka Z. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin 2016;32:829-834
[2] Pineda C, Castaneda Hernandez G, Jacobs IA et al. Assessing the Immunogenicity of Biopharmaceuticals. BioDrugs 2016;30:195-206
[3] Multidisciplinary: biosimilar. Informationen der EMA, www.ema.europa.eu
[4] Handbuch Biosimilars 2017. Pro Generika e. V., Arbeitsgemeinschaft Pro Biosimilars, www.probiosimilars.de
[5] Biosimilars in the EU: Information guide for healthcare professionals; www.ema.europa.eu
Autoren
Prof. Dr. Theo Dingermann ist Seniorprofessor am Institut für Pharmazeutische Biologie an der Goethe-Universität Frankfurt.
Dr. Ilse Zündorf ist dort als akademische Oberrätin tätig.
Institut für Pharmazeutische Biologie, Biozentrum,
Max-von-Laue-Straße 9, 60438 Frankfurt/Main



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.