

















- DAZ.online
- DAZ / AZ
- DAZ 41/2017
- MTX und Co. bei Rheuma

Foto: Neissl – stock.adobe.com
Thema Rheuma
MTX und Co. bei Rheuma
Neue und bewährte Optionen in der Therapie der rheumatoiden Arthritis
Grundlagen der rheumatoiden Arthritis
Die rheumatoide Arthritis (RA) ist eine autoimmun-indizierte chronische Entzündungserkrankung der Synovialmembran, das heißt der Gelenkinnenhaut, die zu einer destruierenden Gelenkveränderung führt. Die Erkrankung ist durch eine schubförmig auftretende Entzündung in den betroffenen Gelenken gekennzeichnet, die mit starken Schmerzen einhergehend unbehandelt progressiv zum Funktionsverlust führt. Wie auch bei anderen autoimmunen Entzündungserkrankungen ist die eigentliche Ursache des fehlgeleiteten Immunsystems nicht bekannt, die Pathogenese der Erkrankung bietet aber verschiedene Ansatzpunkte für eine, für die Patienten lebenslang notwendige Pharmakotherapie. Die rheumatoide Arthritis ist eine systemische Erkrankung, bei ca. 50% der Patienten manifestiert sie sich auch in extraartikulären Pathologien, unter anderem in kardiovaskulären Erkrankungen, subkutanen Gewebeveränderungen (Rheumaknoten), Erschöpfungszuständen sowie depressiven Verstimmungen. Frauen sind von dieser Erkrankung mehr als doppelt so häufig wie Männer betroffen, das Erkrankungsalter gipfelt zwischen dem 55. und 65. Lebensjahr.
Mit einer Inzidenz von ca. 0,8% in Mitteleuropa kann die rheumatoide Arthritis als eine Volkskrankheit klassifiziert werden, die in ihren Auswirkungen nicht nur für die betroffenen Patienten schwerwiegend ist, sondern auch im gesamtgesellschaftlichen Kontext durch direkte (Behandlung) und indirekte Kosten (Verrentung, Arbeitsunfähigkeit) einen hohen Stellenwert besitzt.
Basistherapie
Die Pharmakotherapie der rheumatoiden Arthritis zielt grundsätzlich auf eine Verhinderung der Krankheitsprogression und der damit einhergehenden Gelenkdestruktion, auf die Linderung der Schmerzen sowie auf eine Abschwächung der Entzündungsaktivität. Entsprechend der therapeutischen Leitlinien der European League Against Rheumatism (EULAR) sollte schnellstmöglich nach Diagnostizierung einer rheumatoiden Arthritis mit einer Therapie begonnen werden. In der First-line-Behandlung kommen hier die sogenannten Basistherapeutika zum Einsatz. Diese Wirkstoffgruppe umfasst einige sehr heterogene Arzneistoffe, die längerfristig die Krankheitsprogression verhindern und daher als Disease Modifying Antirheumatic Drugs (DMARD) bezeichnet werden. DMARDs weisen aber keine analgetische Wirkkomponente auf. Der „Goldstandard“ der DMARDs ist der Folsäure-Antagonist Methotrexat (MTX). Leitliniengerecht wird eine Therapie mit oralem MTX begonnen. Unter Umständen kann zur Überbrückung bis zum Wirkungseinsatz niedrig dosiertes Prednisolon oder, symptomatisch fokussiert, die kurzfristige Gabe von COX-Hemmern ergänzt werden.
Zeigt sich nach vier bis acht Wochen kein therapeutisches Ansprechen, kann die orale Methotrexat-Dosis erhöht oder Methotrexat subkutan appliziert werden. Bei ca. 30% der Erkrankten tritt hierdurch keine ausreichende Besserung ein. Dann kann Methotrexat mit einem anderen DMARD ergänzt werden (primär Leflunomid, oder Hydrochloroquin mit Sulfasalazin). Bei anhaltend hoher Krankheitsaktivität oder MTX-Unverträglichkeit können nach ca. drei Monaten weitere therapeutische Optionen hinzugezogen werden.
Therapeutische Optionen jenseits der Basistherapie
Bei Versagen der Basistherapie mit zwei verschiedenen DMARDs bzw. bei deren Unverträglichkeit kann mittlerweile auf ein breites Spektrum neuartiger therapeutischer Prinzipien zurückgegriffen werden. Dabei fokussieren die meisten Wirkstoffe auf die direkte Hemmung proinflammatorischer Zytokine, sowie neuerdings auch auf deren Rezeptoraktivität und die damit verbundene Signalwirkung. Weitere Strategien zielen auf die Aktivierung der T-Zellen sowie die immunologische Eliminierung von B-Lymphozyten. Aus therapeutischer Sicht stellt dies aber keine Rangfolge dar. Diese pharmakologischen Strategien werden grundsätzlich als nahezu gleichwertig eingestuft, die Auswahl kann unter Umständen vom Nebenwirkungsprofil der Wirkstoffe oder Komorbiditäten der Patienten bestimmt werden.
Nebenwirkungen und Kontraindikationen der TNF-α-Blockade
Die Blockade des TNF-α verursacht eine Immunsuppression der Patienten. Bestimmte Vorsichtsmaßnahmen, Überwachungen und Kontraindikationen bei Therapie mit TNF-α-Inhibitoren sind zu berücksichtigen:
- Nebenwirkungen: Infusionsbedingte (anaphylaktische) Reaktionen, erhöhte Infektanfälligkeit (insbesondere des Respirationstrakts, besonderes Augenmerk wird auf Tuberkulose gelegt), Reaktivierung von viralen Infekten, invasive Pilzinfektionen, Bildung von Autoantikörpern, eine beschriebene Lymphomanfälligkeit ist aufgrund der geringen Fallzahlen nicht signifikant zu bewerten.
- Kontraindikationen: Aktive Infekte, Tuberkulose sowie andere immunsuppressive Therapien sind Kontraindikationen, schwere bis mittelschwere Herzinsuffizienz schließen ebenfalls eine Therapie aus.
Hemmung proinflammatorischer Zytokine
Zytokine sind eine große Gruppe von Mediatorstoffen der zellulären Kommunikation. Im Kontext der Entzündung – und im engeren Sinne der Therapie der rheumatoiden Arthritis – spielen hierbei die proinflammatorischen Interleukine IL-1, IL-6 sowie der Tumornekrosefaktor alpha (TNF-α) eine herausragende Rolle. Mit deren Hemmung wird in verschiedene Schritte der autoimmun-induzierten Überaktivierung der Immunzellen eingegriffen. Während TNF-α und IL-6 gegenwärtig als attraktive Targets in der Therapie der rheumatoiden Arthritis adressiert werden, ist IL-1 aktuell für diese Indikation fast bedeutungslos. Der rekombinante IL-1 Rezeptorantagonist Anakinra (Kineret®) wurde 2002 für die Therapie der rheumatoiden Arthritis in Kombination mit MTX zugelassen, besitzt aber aufgrund seiner notwendig täglichen Applikation keine Bedeutung mehr für diese Therapie.
TNF-α-Hemmung als antiinflammatorische Strategie
Der Tumornekrosefaktor alpha ist als ein zentraler Mediator in vielfältige pathologische Entzündungsreaktionen und Prozesse involviert, so dass eine Hemmung dieses Zytokins eine kausale Blockade des Entzündungsfortgangs auch in der Therapie der rheumatoiden Arthritis ermöglicht. Aktuell existieren fünf rekombinante Wirkstoffe auf dem Arzneimittelmarkt, die als TNF-α-Hemmer trotz ihrer strukturellen Diversitäten untereinander (Abb. 1) als therapeutisch gleichwertig einzuschätzen sind. Soll die Basistherapie der rheumatoiden Arthritis ergänzt werden, fällt die primäre Entscheidung zumeist auf den Einsatz eines TNF-α-Blockers.
Infliximab (Remicade®) wurde als der erste Wirkstoff zur Blockade des TNF-α bereits 1999 zugelassen. Infliximab ist ein chimärer (murin/humaner) IgG1-Antikörper gegen TNF-α, der in murinen Hybridom-Zellen hergestellt wird. Der Erstzulassung zur Therapie des Morbus Crohn folgte unter anderem auch die Indikationserweiterung zur Therapie der rheumatoiden Arthritis in Kombination mit Methotrexat für solche Patienten, die nicht ausreichend auf zwei DMARDs ansprechen.
Therapie mit Infliximab
Infliximab wird intravenös über ca. zwei Stunden in Kombination mit Methotrexat in einer Dosierung von 3 mg/kg Körpergewicht bei Therapiebeginn sowie nach zwei und sechs Wochen verabreicht; das folgende Applikationsintervall beträgt dann acht Wochen. Der Wirkungseintritt ist in der Regel nach zwei bis drei Wochen sichtbar. Mit dem Wegfall des Patentschutzes des Remicade® im Februar 2015 wurden zwei Biosimilars, die zueinander „bioidentischen“ (gleiche Produktionslinie) Remsina® und Inflectra® auf dem europäischen Arzneimittelmarkt zugelassen, gefolgt vom dritten Biosimilar Flexabi® im Jahr 2016.
Adalimumab (Humira®) ist ein humaner IgG1-Antikörper gegen TNF-α, der in CHO-Zellen generiert wird. Seit dem Jahr 2003 ist Adalimumab zur Therapie der rheumatoiden Arthritis (Abbvie) zugelassen, vielfältige Indikationserweiterungen folgten, unter anderem auch zur Behandlung der polyartikulären juvenilen Arthritis (ab vier Jahre). Im Gegensatz zu Infliximab kann wegen des geringeren immunogenen Potenzials des humanen Antikörpers Adalimumab auch als Monotherapie verwendet werden. Wegen der vielfältigen Indikationsgebiete für Adalimumab und der breiten Anwendung wurde der Antikörper zum umsatzstärksten Arzneimittel der letzten Jahre weltweit. Im März 2017 erhielten die beiden Biosimilars Amgevita® und Solymbic® (Amgen, zueinander bioidentisch) die Zulassung in Europa.
Therapie mit Adalimumab
Adalimumab wird als subkutane Injektion von 40 mg im zweiwöchigen Abstand appliziert. Die bereits genannten Nebenwirkungen der Anwendung sowie Einschränkungen durch Immunsuppression der Patienten sind zu beachten. Obwohl die humane Antikörperstruktur ein offensichtlich geringeres immunogenes Risiko beinhaltet, tritt bei ca. 30% der Patienten ein Wirkungsverlust in der Therapie auf, der mit der Bildung von Antikörpern gegen den Wirkstoff assoziiert ist.
Etanercept (Enbrel®) ist ein weiterer TNF-α-Antagonist. Strukturell handelt es sich um ein Dimer eines chimären Proteins, welches aus der extrazellulären Ligandbindungsdomäne des humanen TNF-α-Rezeptors 2 und der Fc-Domäne eines humanen IgG1 besteht. Etanercept kann somit zwei Moleküle TNF-α binden. Es unterscheidet sich in zweierlei Hinsicht von den oben genannten Antikörpern: es bindet nur freies, nicht aber membranär gebundenes TNF-α. Und Etanercept kann auch Lymphotoxin A (TNF-β) binden. Daher weist Etanercept ein etwas anderes Wirkungs- und Nebenwirkungsprofil als die TNF-α-Antikörper auf. Die Zulassung (Wyeth) erfolgte bereits 2000 zur Therapie der rheumatoiden Arthritis bei Versagen der Basistherapie, es folgten verschiedene Indikationserweiterungen einschließlich der juvenilen idiopathischen Polyarthritis (ab zwei Jahre).
Therapie mit Etanercept
Etanercept wird subkutan in einer Dosierung von 25 mg zweimal wöchentlich appliziert. Etanercept wird als gut verträglich eingeschätzt, der Wirkungseintritt ist bereits nach ca. zwei Wochen zu erwarten, die resultierende Immunsuppression erfordert aber auch die genannten Vorsichtsmaßnahmen und Berücksichtigung von Kontraindikationen. Etanercept ist ebenfalls unter den Top Five der umsatzstärksten Arzneimittel weltweit zu finden.
Mit Benepali® wurde im Januar 2016 das erste Biosimilar des Enbrel® in Europa zugelassen, Erelzi® (Sandoz) folgte im Juni 2017 als zweites Nachahmerpräparat.
Golimumab (Simponi®) ist ein humaner IgG1-Antikörper gegen TNF-α, der in einer murinen Hybridom-Zelllinie hergestellt wird. Golimumab erhielt 2009 (Centocor B.V.) die Zulassung zur Therapie der rheumatoiden Arthritis in Kombination mit MTX.
Therapie mit Golimumab
Der Antikörper wird in einer Dosierung von 50 mg einmal monatlich subkutan durch eine Fertigspritze mittels eines Autoinjektors durch die Patienten selbst verabreicht. Hinsichtlich Nebenwirkungen und Kontraindikationen gelten die genannten Aspekte der immunsuppressiven Wirkung des Antikörpers.
Certolizumab Pegol (Cimzia®) ist ein weiterer TNF-α-Antagonist, der für die Therapie der rheumatoiden Arthritis bei Versagen der Standardbehandlung in Kombination mit MTX seit 2009 zugelassen ist (UCB Pharma). Certolizumab Pegol ist ein in Escherichia coli gebildetes humanisiertes Antigen-bindendes (Fab-)Fragment eines TNF-α-Antikörpers. Zur Verlängerung seiner Zirkulationszeit wurde das Fragment mit einer Polyethylenglykol-Kette verbunden. Der Einsatz eines Antikörperfragments anstelle der kompletten Antikörperstruktur zielt auf die Umgehung des immunogenen Fc-Bereiches für eine verminderte Komplementaktivierung sowie reduzierte antikörperabhängige zelluläre Zytotoxizität.
Therapie mit Certolizumab Pegol
Certolizumab Pegol wird als subkutane Injektion im zweiwöchigen Abstand in einer Dosierung von 400 mg, ab der dritten Gabe 200 mg verabreicht. Trotz der vereinfachten Antikörperstruktur unterscheidet sich Certolizumab Pegol nicht deutlich in Wirkung und Nebenwirkung von den anderen TNF-α-Antagonisten.
Stellenwert der TNF-α-Antagonisten
Die TNF-α-Antagonisten waren die ersten rekombinanten Proteinarzneistoffe, die in der Therapie der rheumatoiden Arthritis ein neues Zeitalter eingeläutet haben. Erstmals gelang es durch die Wirkstoffgruppe, bei den mittels DMARD nicht mehr ausreichend therapierbaren Patienten den Erkrankungsfortschritt signifikant einzudämmen, so dass nicht das Verlangsamen, sondern das Aufhalten der Gelenkdestruktionen therapeutisch greifbar wurde. Obwohl die Anwendung der TNF-α-Blocker zweifelsohne als die Primärstrategie bei Versagen der Standardtherapie erscheint, und dies auch die starke Akzeptanz dieser Wirkstoffe und auch deren ökonomischen Erfolg begründet, spricht doch ein relativ hoher Anteil der Patienten mit schweren Verlaufsformen (ca. 30%) nicht, oder nach erfolgter Therapie nicht mehr auf diese Intervention an. Für diese Patienten wurden neue therapeutische Optionen eröffnet.
Beratung zu TNF-α-Antagonisten in der Apotheke
Die breite Akzeptanz der TNF-α-Antagonisten bringt diese Wirkstoffe nahezu täglich in die Apothekenpraxis. Daraus ergeben sich für die Beratung der Patienten vielfältige Ansatzpunkte. Durch die resultierende Immunsuppression gilt der Selbstmedikation der Patienten besondere Aufmerksamkeit. Insbesondere für die Reiseberatung muss die Immunsuppression berücksichtigt werden. So sollte auf Gefahren durch Infektionen mit bakteriellen (Tuberkulose!) oder parasitären Erregern gesondert hingewiesen werden. Impfungen mit Totimpfstoffen gelten in der Regel für diese Patienten als komplikationslos, Impfungen mit Lebendimpfstoffen müssen durch den behandelnden Arzt erwogen werden.
Wie bei allen Biologicals sind durch den Protein-Charakter der Wirkstoffe hinsichtlich der Metabolisierung kaum Wechselwirkungen mit anderen Arzneistoffen zu beachten (s. u.).
Ratschläge für die richtige Anwendung sollten insbesondere
- die langsame Erwärmung der Produkte zur Applikation nach vorgeschriebener Kühlschranklagerung beinhalten,
- das Vermeiden des Schüttelns der Applikatoren einschließen sowie
- auf das Kühlen der Einstichstelle zur Verminderung lokaler Reaktionen hinweisen.
Hemmung von Interleukin 6
Tocilizumab zur Hemmung des IL-6-Rezeptors. Interleukin 6 (IL-6) ist in verschiedene Prozesse der pathologischen Progression bei der rheumatoiden Arthritis kritisch involviert, z. B. Migration und Aktivierung von T- und B-Lymphozyten sowie Aktivierung von Osteoklasten. Tocilizumab (RoActemra®) ist ein humanisierter Antikörper, der sowohl die lösliche als auch membrangebundene Form des IL-6-Rezeptors bindet und so IL-6 in seiner proinflammatorischen Wirkung hemmt. Der Antikörper ist seit 2009 als Infusion mit MTX zur Therapie Erwachsener mit schwerer progressiver rheumatoider Arthritis zugelassen, seit 2014 kann Tocilizumab auch subkutan als Monotherapie bei Patienten mit mäßiger bis mittelschwerer RA angewendet werden.
Therapie mit Tocilizumab
Tocilizumab wird als einstündige Infusion (20 mg/ml) in einer Dosierung von 8 mg/kg Körpergewicht alle vier Wochen intravenös appliziert; für die subkutane Anwendung gilt eine feste Dosierung von 162 mg wöchentlich. Auch für Tocilizumab gelten die vorab genannten Begleiterscheinungen und Kontraindikationen der resultierenden Immunsuppression der Patienten.
Sarilumab zur Hemmung des IL-6-Rezeptors. Seit August 2017 existiert mit Sarilumab (Kevzara®) ein weiterer Wirkstoff für erwachsene Patienten mit einer mittelschweren bis schweren rheumatoiden Arthritis, die auf ein oder mehrere DMARDs unzureichend angesprochen oder diese nicht vertragen haben. Sarilumab ist ein humaner Antikörper gegen den IL-6-Rezeptor, der somit den gleichen Wirkmechanismus wie Tocilizumab aufweist und mit Methotrexat, aber auch als Monotherapie angewendet wird. In der Zulassungsstudie war Sarilumab in seiner Wirkung dem Adalimumab überlegen.
Therapie mit Sarilumab
Sarilumab wird in einer Dosierung von 200 mg subkutan alle zwei Wochen appliziert, dies kann nach einer entsprechenden Schulung durch den Patienten selbst erfolgen. Bei Neutropenie, Thrombozytopenie, erhöhten Leberenzymwerten oder schweren Infektionen soll die Dosis auf 150 mg alle zwei Wochen gesenkt, die Behandlung gar nicht erst begonnen, unterbrochen oder beendet werden.
Die häufigsten Nebenwirkungen in Studien waren Neutropenie, erhöhte Werte des Leberenzyms Alanin-Aminotransferase (ALT), Infektionen der oberen Atemwege und Harnwegsinfekte. Unter den schwerwiegenden Nebenwirkungen waren Infektionen, insbesondere Pneumonien am häufigsten.
Blick in die Pipeline
Durch die überzeugende Wirksamkeit der IL-6-Hemmung, insbesondere für Patienten die nicht adäquat auf TNF-α-Inhibition ansprechen, befinden sich verschiedene IL-6-Antikörper in der klinischen Entwicklung.
Vobarilizumab ist ein weiterer Antikörper gegen den IL-6-Rezeptor. Der Antikörper wird von Ablynx entwickelt und befindet sich aktuell in Phase-IIb-Studien für RA-Patienten, die nicht auf MTX ansprechen.
Sirukumab ist ein humaner Antikörper gegen IL-6, der nach positiven Phase-III-Studien zur Therapie der rheumatoiden Arthritis [1] von den Firmen GSK und Janssen bei EMA und FDA zur Zulassung eingereicht wurde. Im August 2017 hat allerdings der Zulassungsausschuss der FDA wegen Sicherheitsbedenken keine Zulassungsempfehlung ausgesprochen.
Clazakizumab ist ein in Hefezellen produzierter, aglykosylierter humanisierter Antikörper gegen IL-6, der gegenwärtig durch Vitaeris entwickelt eine klinische Studie IIb für die Therapie der rheumatoiden Arthritis [2] erfolgreich abgeschlossen hat.
Olokizumab ist ein humanisierter IL-6-Antikörper, der entwickelt durch UCB Pharma im Jahre 2016 erfolgreiche Phase-II-Studienergebnisse zur Behandlung von Patienten mit rheumatoider Arthritis zeigte, er war sogar in der Wirkung gegenüber TNF-α überlegen [3].
IL-6-Hemmung und CYP-Interaktionen
Obwohl die rekombinanten Proteine kaum Anlass für eine Wechselwirkung mit der Biotransformation anderer Arzneistoffe geben, gilt dies für die beiden IL-6-Rezeptor-Antagonisten nicht. Da IL-6 verschiedene Subenzyme des CYP450-Systems unterdrückt, können die beiden Antikörper Tocilizumab und Sarilumab durch ihre Interferenz auf Ebene des IL-6 das Enzymsystem in seine „normale“ Aktivität zurück überführen und somit die Wirkstoffspiegel anderer Arzneistoffe (z. B. Atorvastatin, Simvastatin, Warfarin, Ciclosporin, Phenytoin) beeinflussen.
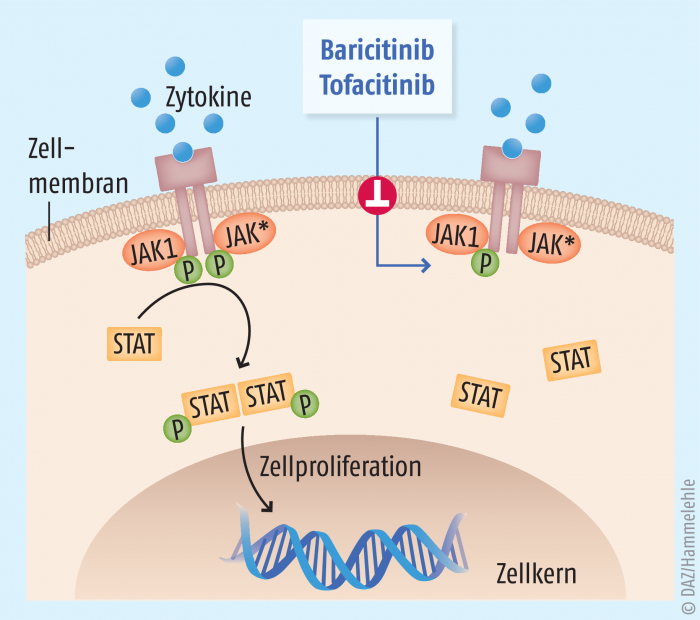
Hemmung der Signalkaskaden proinflammatorischer Zytokine durch Januskinase-Inhibitoren
Proinflammatorische Zytokine vermitteln ihre Entzündungsreaktion durch spezifische Rezeptoren auf den unterschiedlichen Immunzellen. Auch hier werden, wie bei vielen intrazellulären Signalkaskaden, eine Reihe von Phosphorylierungsreaktionen durch Kinasen angestoßen. Allerdings sind die Zytokin-Rezeptoren selbst keine Kinasen, so dass sie für die Signalweiterleitung auf assoziierte Kinasen angewiesen sind. Diese, jeweils paarweise an einem Rezeptor fungierenden Enzyme, werden als Januskinasen (JAK) bezeichnet. Die Familie der Januskinasen umfasst vier Mitglieder; JAK1 bis 3 sowie Tyk2. Diese wiederum vermitteln ihre Wirksamkeit über sogenannte STAT-Proteine (Signaltransduzierer und Aktivator der Transkription) zur Transkriptionsaktivierung im Zellkern (Abb. 2).
Die Inhibierung dieser Januskinase-Paare eröffnet also eine neue therapeutische Option, hemmend in den Entzündungsfortgang einzugreifen. Vorteilhaft erscheint hierbei die Tatsache, dass so nicht nur einzelne Zytokine gehemmt werden, sondern unter Umständen auch die Signalkaskade mehrerer Zytokine gleichzeitig.
Januskinase
Die Namensgebung für diese Rezeptorfamilie erscheint im Kontext der vielfältigen Kinasen des menschlichen Organismus etwas überraschend. Die bei ihrer „Entdeckung“ als weitere neue Kandidaten der großen Kinasefamilie (> 500) simpel als „Just Another Kinase“ bezeichneten Enzyme erhielten erst sekundär die Assoziation zum römischen Gott Janus, dem Hüter von Ein- und Ausgang. Diese Verbindung passt allerdings für die Vermittlerfunktion der Januskinasen perfekt. Für die betreffenden Hemmstoffe der Januskinasen hat sich die Bezeichnung „Jakinibe“ etabliert.
Mit Baricitinib (Olumiant®) wurde im April 2017 der erste Januskinase-Hemmstoff in Europa in die Therapie der rheumatoiden Arthritis eingeführt [4]. Baricitinib ist ein selektiver Inhibitor von JAK1 und JAK2 und hemmt damit nicht nur die Signalkaskade des IL-6, sondern auch die der Interferone α/β. Baricitinib ist zur Behandlung erwachsener Patienten mit mittelschwerer bis schwerer rheumatoider Arthritis als Monotherapie oder Kombination mit Methotrexat zugelassen, die auf eine vorangegangene Behandlung mit einem oder mehreren DMARDs unzureichend angesprochen oder diese nicht vertragen haben. In den Zulassungsstudien zeigte Baricitinib eine Wirkungsüberlegenheit gegenüber Methotrexat und Adalimumab.
Therapie mit Baricitinib
Olumiant® Filmtabletten werden, unabhängig von Tageszeit oder Mahlzeiten in einer Dosis von 4 mg täglich (einmalig oder aufgeteilt in zwei 2 mg Einnahmen) angewendet. Für ältere Patienten (> 75 Jahre) sollte die geringere 2-mg-Dosis gewählt werden. Dies gilt auch für Patienten mit eingeschränkter Nierenfunktion sowie bei der gleichzeitigen Einnahme von Probenecid. Dieser kann als Inhibitor des Organischen Anionen-Transporters Typ 3 die Pharmakokinetik von Baricitinib beeinflussen. Nicht empfohlen wird die Anwendung bei Patienten mit schwerer Leberfunktionsstörung und einer Kreatinin-Clearance unter 30 ml/Minute.
Als Nebenwirkungen sind das Auftreten von Hypercholesterolämie sowie Infektionen der oberen Atemwege, virale Infektionen wie Gürtelrose sowie weiterhin Übelkeit und Harnwegsinfektionen beschrieben. Die erhöhte Infektionsrate ist Ausdruck der Immunsuppression, daher müssen vor Therapiebeginn die Patienten auf Tuberkulose getestet werden. Bei Patienten mit chronischen oder wiederkehrenden Infektionen sollten ebenfalls vor Therapiebeginn Nutzen und Risiken der Therapie abgewogen werden. Baricitinib darf nicht in der Schwangerschaft angewendet werden.
Mit Tofacitinib (Xeljanz®) kam im Mai 2017 ein weiterer Januskinase-Hemmer auf den europäischen Arzneimittelmarkt für die Therapie von Patienten mit mittelschwerer oder schwerer rheumatoider Arthritis, die nicht auf eine Therapie mit DMARDs ansprechen [5]. Interessanterweise ist Tofacitinib seit 2012 für diese Therapie in den USA zugelassen, eine Zulassungsempfehlung der EMA wurde zu diesem Zeitpunkt nicht ausgesprochen. Tofacitinib hemmt bevorzugt solche Rezeptoren, an denen JAK1 und/oder JAK3 beteiligt sind. Dadurch wird insbesondere die Signalübertragung von IL-2, -4, -6, -9, -15 und -21 sowie von Typ-I- und Typ-II-Interferonen reduziert.
Therapie mit Tofacitinib
Tofacitinib soll in einer Dosierung von zweimal täglich 5 mg, unabhängig von den Mahlzeiten angewendet werden. Bei einer Kreatinin-Clearance < 30 ml/Minute oder einer mittelschweren Leberfunktionsstörung soll die Dosierung auf einmal täglich 5 mg reduziert werden. Die häufigsten Nebenwirkungen einer Therapie mit Tofacitinib waren Kopfschmerzen, Infektionen der oberen Atemwege, Nasopharyngitis, Übelkeit sowie Durchfall.
Unterdrückung der T-Zell-Aktivierung durch „Kostimulationshemmer“
T-Lymphozyten werden zur Aktivierung ihrer immunologischen Abwehrfunktion durch Antigene spezifisch funktionalisiert. Diese physiologische Abwehrfunktion kann aber bei autoimmunen Entzündungserkrankungen wie der rheumatoiden Arthritis durch Autoantigene entkoppelt sein und zum Erkrankungsfortgang entscheidend beitragen. Der Einblick in die molekularen Mechanismen der T-Zell-Aktivierung hat neue therapeutische Ansatzpunkte eröffnet.
Der Prozess der Antigenpräsentation durch Antigen-präsentierende Zellen (APC) unterliegt dabei einem komplexen Erkennungsmechanismus mit doppeltem Kontrollsystem. Das von der Zelle präsentierte Antigen wird durch den T-Zell-Rezeptor erkannt, aber erst ein weiteres Kostimulationssignal von CD80/CD86 auf die Antigen-präsentierenden Zellen mit CD28 auf den T-Zellen führt zu deren Aktivierung. Für eine Autoregulation exprimieren aktivierte T-Zellen dann das zytotoxische T-Lymphozyten-assoziierte Protein 4 (CTLA-4), welches als Gegenspieler der Kostimulation mit deutlich höherer Affinität als CD28 an CD80/CD86 bindet und so die Stimulation unterbindet (Abb. 3).
Durch rekombinantes CTLA-4 kann daher pharmakologisch hemmend in die T-Zell-Aktivierung für eine Eindämmung der autoimmunen Entzündungen eingegriffen werden.
Mit Abatacept (Orencia®, Bristol-Myers Squibb) wurde bereits im Jahre 2007 ein rekombinantes CTLA-4-Analogon zur Therapie erwachsener Patienten mit mäßigem oder schwerem Krankheitsverlauf zugelassen (in Kombination mit MTX), wenn die Patienten nicht auf die entsprechende Standardtherapie mit DMARDs ansprechen. 2010 wurde die Zulassung auch für Kinder ab sechs Jahren mit juveniler idiopathischer Arthritis nach Versagen der DMARDs oder einem TNF-α-Blocker erweitert. Abatacept ist ein lösliches Fusionsprotein, das aus der extrazellulären Domäne des humanen CTLA-4 sowie dem modifizierten Fc-Bereich eines humanen IgG1-Antikörpers besteht und in CHO-Zellen produziert wird. Abatacept liegt, über eine Disulfid-Brücke verknüpft, als Homodimer vor, was offensichtlich die Bindungsfähigkeit an CD80/CD86 verstärkt und so die Deaktivierung der T-Zellen verbessert.
Therapie mit Abatacept
Der Wirkstoff wird in einer Dosierung von 10 mg/kg Körpergewicht als halbstündige Infusion an Tag 1, 15 und 30 gefolgt von monatlichen Infusionen appliziert. Als Nebenwirkungen sind Kopfschmerzen, Benommenheit und erhöhter Blutdruck beschrieben. Durch die resultierende Immunsuppression sind auch bei dieser Therapie wiederum erhöhte Infektanfälligkeiten, insbesondere Atemwegsinfektionen als typische Nebenwirkungen zu beobachten. Opportunistische Infektionen, virale Hepatitis oder Tuberkulose sind Kontraindikationen, eine Kombination mit TNF-α-Blockern ist zu vermeiden.
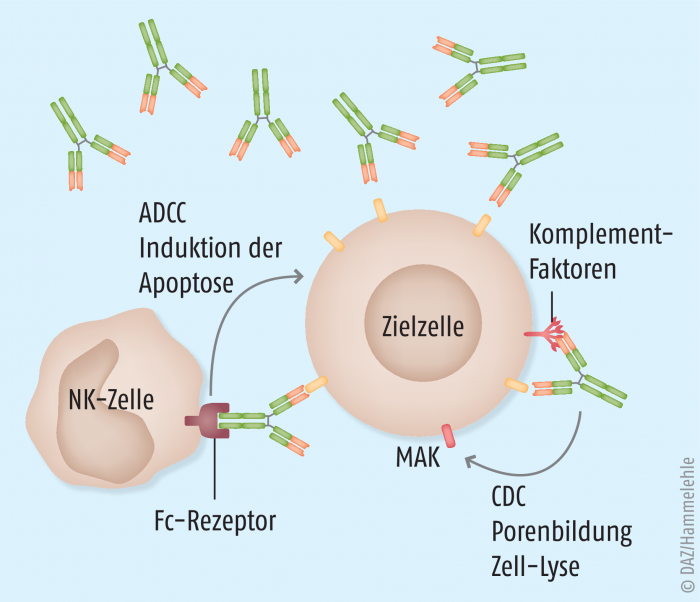
Eliminierung überreaktiver B-Zellen als therapeutische Option
Die Grundlage dieser therapeutischen Strategie liegt in der immunologischen Funktion von Antikörpern, die nach Bindung an pathologischen Strukturen das Immunsystem auf die so markierten Elemente richten. Hierbei kommen dann, wie in Abbildung 4 dargestellt, die Prozesse der zellulären und humoralen Immunabwehr (Komplementsystem) funktionell zusammen und sorgen für eine Eliminierung der pathogenen Struktur. Dieses Eliminierungsprinzip kann auch auf körpereigene Zellen gerichtet werden, wenn diese durch spezifische Antikörper markiert werden.
Seit vielen Jahren wird diese Strategie zur Bekämpfung maligner Leukozyten in der Therapie leukämischer Erkrankungen erfolgreich angewendet. Da aber auch bei autoimmunen Entzündungserkrankungen überschießende Immunzellen entscheidend zur Pathogenese beitragen, eignet sich dieses Therapieprinzip gleichermaßen. CD20, ein von B-Zellen und deren späten Vorstufen spezifisch exprimiertes Glykoprotein (B-lymphocyte antigen) wird beispielsweise adressiert, um B-Lymphozyten zu eliminieren. Somit können bei pathologischen Entzündungserkrankungen mit starker B-Zell-Involvierung – neben rheumatoider Arthritis auch bei der multiplen Sklerose – antientzündliche Effekte erzielt werden. Da CD20 nicht auf frühen Vorläuferzellen der B-Zell-Population sowie Stammzellen exprimiert wird, werden diese Zellen nicht erfasst und können nach erfolgter therapeutischer Intervention für eine Regenerierung der B-Zellen sorgen.
Rituximab. Rituximab (MabThera®, Roche) ist ein in CHO-Zellen hergestellter monoklonaler, murin/human-chimärer CD20 Antikörper, der bereits 1998 die Zulassung zur Therapie des follikulären Non-Hodgkin-Lymphoms erhielt. Seit dem Jahr 2006 ist Rituximab auch zur Therapie der rheumatoiden Arthritis in Kombination mit Methotrexat zugelassen, wenn die Patienten nicht ausreichend auf eine Basistherapie sowie den Einsatz von TNF-α-Blockern ansprechen oder diese vertragen.
Therapie mit Rituximab
MabThera® wird in einer Packungsgröße von 100 mg angeboten, der Antikörper wird in einer Konzentration von 10 mg/ml i. v. angewendet. Für die Therapie der rheumatoiden Arthritis werden zweimal je 1000 mg im Abstand von zwei Wochen appliziert, nach Ansprechen der Therapie nach ca. ein bis drei Wochen wird unter Umständen nach 24 Wochen eine weitere Injektion vorgenommen.
Rituximab wird generell gut vertragen; beschriebene Nebenwirkungen beziehen sich hauptsächlich auf infusionsbedingte Reaktionen. Zur Kontrolle von infusionsbedingten Nebenwirkungen, insbesondere dem sogenannten Zytokin-Freisetzungs-Syndrom, sind die Patienten engmaschig zu überwachen. Es ist auf eine langsame Infusionsrate (anfangs 50 mg/Stunde) zu achten; eine Prämedikation (Analgetikum/Antihistaminikum) ist anzuwenden.
Die mit der Therapie einhergehende Immunsuppression erfordert bestimmte Vorsichtsmaßnahmen. Daher sind bestehende aktive schwere Infektionen oder bereits immunsupprimierte Patienten klare Gegenanzeigen einer Therapie. Vor einer Therapie mit Rituximab muss zum Ausschluss einer Reaktivierung von Hepatitis B ein Virusscreening (Rote-Hand-Brief 11/2013 [6]) durchgeführt werden. Zudem ist zum Ausschluss einer sich entwickelnden progressiven multifokalen Leukenzephalopathie (PML) eine engmaschige Überwachung der Patienten auf JC-Virustiter notwendig.
Mittlerweile existieren auch zwei Biosimilars des MabThera® auf dem europäischen Markt. Im Februar 2017 wurden Truxima® (Celltrion; sowie das Bioidentical Ritemvia®) und im Juni 2017 Rixathon® und das Bioidentical Riximyo®(Sandoz) zum Indikationsspektrum des Originators einschließlich Therapie der rheumatoiden Arthritis zugelassen.
Wertung
Für die Therapie der schweren Verlaufsformen der rheumatoiden Arthritis existiert mittlerweile ein breites Wirkstoff- sowie Therapiespektrum zur Ergänzung der DMARDs bei deren Nichtwirksamkeit oder Unverträglichkeit. Obwohl diese neuen Therapieprinzipien nichts an der grundsätzlichen Unheilbarkeit der autoimmunen Erkrankung ändern können, ermöglichen sie mit beeindruckenden antiinflammatorischen Wirkungen den Patienten ein Leben mit dieser Erkrankung in nahezu normaler Aktivität sowie eine Stagnation des Krankheitsfortganges. Die Vielfältigkeit der therapeutischen Interventionen eröffnet die Möglichkeit, bei Unverträglichkeiten oder unzureichenden Wirksamkeiten auf alternative Ansätze auszuweichen. Zu berücksichtigen bleibt aber, dass die genannten Prinzipien mit einer Immunsuppression der Patienten sensitiv in die immunologische Balance eingreifen, was einer hohen Aufmerksamkeit für die therapeutische Begleitung der Patienten bedarf und so auch hohe Anforderungen an die Beratungstätigkeit durch die Apotheker stellt. |
Literatur
[1] Lazzerini PE et al. Spotlight on sirukumab for the treatment of rheumatoid arthritis: the evidence to date. Drug Des Devel Ther 2016;10:3083-3098
[2] Weinblatt ME et al. The efficacy and safety of subcutaneous clazakizumab in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol 2015;67:2591-600
[3] Genovese MC et al. Efficacy and safety of olokizumab in patients with rheumatoid arthritis with an inadequate response to TNF inhibitor therapy: outcomes of a randomised Phase IIb study. Ann Rheum Dis 2014;73:1607-1615
[4] Baricitinib EMA Zulassung: www.ema.europa.eu/docs/de_DE/document_library/EPAR_-_Summary_for_the_public/human/004085/WC500223726.pdf
[5] Tofacitinib EMA Dossier: https://ec.europa.eu/health/documents/community-register/2017/20170322137186/anx_137186_de.pdf
[6] MabThera® (Rituximab): Hepatitis-B-Virus-Screening vor Behandlungsbeginn. Rote-Hand-Brief der Roche Pharma AG, November 2013
Autor
Prof. Dr. Gerd Bendas studierte Pharmazie an der Martin-Luther-Universität Halle. Nach anschließender Promotion erfolgte 2000 die Habilitation für das Fachgebiet Pharmazeutische Chemie. Seit 2003 hat er eine Professur für Pharmazeutische Chemie an der Universität Bonn inne.
Literaturtipp
Sie sind teuer und sie versprechen viel: die Biologicals.
Machen Sie sich vertraut mit diesen innovativen Arzneistoffen, indem Sie:
- Grundkenntnisse zu Struktur und Herstellung aufpolieren,
- beispielhafte Indikationen und Targets kennenlernen,
- Konsequenzen für das Handling in der Apotheke verstehen und
- einen Blick in die Pipeline werfen.
Biopharmazeutika liegen im Trend – daten Sie sich up!
Von Gerd Bendas und Martina Düfer
Update Biologicals
Rekombinante Proteine und ihr therapeutischer Einsatz
XII, 85 S., 42 farb. Abb., 2 farb. Tab.,
21,0 x 29,7 cm, kartoniert, 24,80 Euro,
ISBN 978-3-7692-6628-3
Deutscher Apotheker Verlag 2016



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.