

















- DAZ.online
- DAZ / AZ
- DAZ 1/2013
- Prodrug-Hydrolyse von ...
Arzneimittel und Therapie
Prodrug-Hydrolyse von Zytostatikum verhindert
Die Studie wurde kürzlich in Biochemical Pharmacology von Xiao et al. publiziert, die in vitro den hemmenden Einfluss von Orlistat auf Carboxylesterasen (CES) untersuchten [1]. CES sind sowohl als Lipasen aktiv als auch an der Hydrolyse von Medikamenten und Prodrugs beteiligt. Die Autoren fanden eine unerwartet starke Hemmung der an der Arzneistoff-Hydrolyse beteiligten CES und mutmaßten, dass dies klinische Konsequenzen haben könnte. Da Orlistat in Europa seit 2009 bis zu einer Dosis von 60 mg pro Hartkapsel lediglich apothekenpflichtig und erst ab 120 mg verschreibungspflichtig ist, wird es häufig im Rahmen der Selbstmedikation abgegeben. Eine Begleitung der Therapie durch den Arzt ist kaum möglich. Träfen die genannten Interaktionen in vivo zu, könnte die unkontrollierte Einnahme tatsächlich ausgeprägte Interaktionen mit großer klinischer Relevanz bedingen.
Um diese Befunde einschätzen zu können, sollen zunächst kurz die Eigenschaften von Orlistat beleuchtet werden.
Die Wirkung von Orlistat
Orlistat ist ein kovalenter Inhibitor gastrointestinaler Lipasen. Mit der Nahrung aufgenommene Triglyzeride können so nicht in freie Fettsäuren und Monoglyze-ride hydrolysiert werden. Die Folge ist eine verminderte Resorption von Lipiden und Ausscheidung von fettigem Stuhl.
Mit einer Dosisempfehlung von 3 x 60 mg/d ist Orlistat in Verbindung mit einer leicht hypokalorischen Kost zur Behandlung von übergewichtigen Patienten (BMI ≥ 28 kg/m²) mit begleitenden Risikofaktoren indiziert, die Therapie sollte nicht länger als sechs Monate durchgeführt werden. Die rezeptpflichtige 3 x 120-mg-Dosierung ist darüber hinaus bei adipösen Patienten (BMI ≥ 30 kg/m²) indiziert, die in ein Gewichtsreduktionsprogramm eingeschlossen werden.
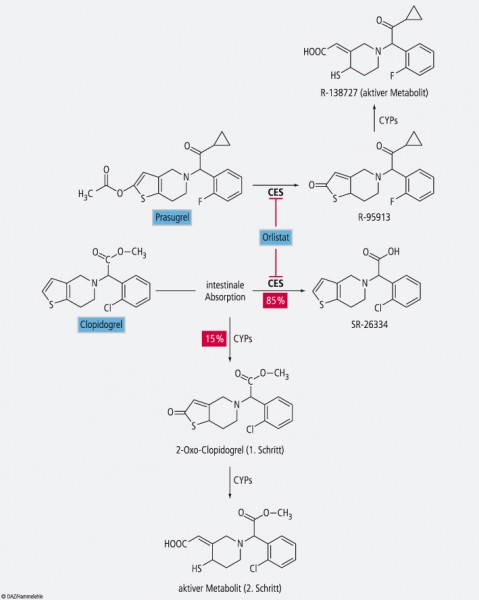
Die Prodrugs Clopidogrel und Prasugrel werden durch hepatische Esterasen (CES) metabolisiert. Eine Orlistat-vermittelte CES2-Hemmung könnte zu erhöhten Wirkspiegeln von Clopidogrel und damit einem erhöhten Blutungsrisiko führen, während Prasugrel nicht in seine aktive Form überführt und das Thromboserisiko steigen würde.
Kein anhaltender Gewichtsverlust
Einer Metaanalyse zufolge trägt Orlistat zur Gewichtsreduktion bei, allerdings gibt es bislang keine Hinweise auf eine Reduktion der Mortalität oder kardiovaskulärer Endpunkte. In 16 Studien mit Orlistat (überwiegend 3 x 120 mg/d), die insgesamt 10.631 Patienten einschlossen, reduzierte Orlistat nach einem Jahr das Gewicht um durchschnittlich 2,9 kg mehr als Placebo [2]. Eine 5%ige Gewichtsreduktion trat bei 21%, eine 10%ige Reduktion bei 12% der Patienten auf. Nach einer Behandlungsdauer von zwei Jahren wurde jedoch kein signifikanter Unterschied mehr zwischen Verum und Placebo beobachtet.
Diabetes-Risiko vermindert
Neben einer leichten Reduktion des Hüftumfangs wurde auch eine leichte Verbesserung der metabolischen Stoffwechsellage beobachtet, Patienten mit gestörter Glucosetoleranz wiesen ein geringeres Risiko für die Manifestation eines Typ-2-Diabetes mellitus auf. Die 3 x 60-mg-Dosierung führt nach sechs Monaten ebenfalls zu einer Reduktion des Gewichtes um ca. 2 kg [2]. Eine längere Behandlung ist hier aber nicht angezeigt.
Problem fettige Stühle
Besonders die fettigen Stühle werden als eine unangenehme Nebenwirkung wahrgenommen – gastrointestinale Störungen sind mit 15 bis 30% sehr häufig – und sind Hauptgrund für einen Therapieabbruch [3]. Hypovitaminosen wurden dagegen interessanterweise kaum beobachtet, obwohl man annehmen könnte, dass diese durch die Steatorrhöen häufiger auftreten würden. Allerdings waren in den beschriebenen Studien alle Patienten angewiesen worden, Multivitaminpräparate einzunehmen. In der Post-marketing-Surveillance sind darüber hinaus zahlreiche Meldungen über hepatobiliäre Ereignisse registriert worden [4].
Wenige Interaktionsstudien
Pharmakokinetische Interaktionsstudien sind rar. Orlistat selbst wird kaum aufgenommen und kann nur in äußerst niedrigen Konzentrationen (<10 ng/ml = 20 nmol/l) im Plasma nachgewiesen werden. Ungefähr 97% der angewendeten Dosis werden mit dem Stuhl ausgeschieden, davon 83% als unverändertes Orlistat. In den Fachinformationen wird auf Wechselwirkungen hingewiesen, die zu einer Reduktion der Bioverfügbarkeit mit anderen Arzneistoffen führen, Ciclosporin ist ausdrücklich kontraindiziert, die Wirkung oraler Antikoagulanzien sollte engmaschig kontrolliert werden [4].
Die Studie zur Interaktion von Orlistat mit Esterasen
Xiao et al. gingen der gängigen Hypothese nach, dass Orlistat die Lipid-hydrolysierende Carboxylesterase 1 (CES1) stärker inhibieren würde als die CES2, die kaum Lipide, aber mehr Arzneistoffe und Prodrugs wie z. B. Irinotecan hydrolysiert [1]. Hierzu untersuchten sie zunächst die Wirkung von Orlistat an Lebermikrosomen aus menschlichem, Maus- und Ratten-Gewebe. Orlistat inhibierte signifikant die humane Esterase-Aktivität bei 1 nmol/l und führte bei 50 nmol/l zu einer 50%igen Hemmung. Auch die CES der Maus konnten bei niedrigen Konzentrationen inhibiert werden, die der Ratte waren dagegen insensitiver. Um die Frage zu lösen, ob die Esterase-Aktivität von der CES1 oder CES2 herrührte, wurden weitere Experimente durchgeführt. Zuerst konnte in einem Assay überraschenderweise eine stärkere Bindung von Irinotecan an die CES2 nachgewiesen werden. Besonders beeindruckend war aber eine Überprüfung in einem rekombinanten Zellsystem, bei dem die einzelnen Esterasen getrennt untersucht werden konnten. Hier führte bereits eine Konzentration von 1 nmol/l Orlistat zur einer 75%igen Inhibition von CES2, eine CES1-Hemmung konnte dagegen nicht nachgewiesen werden. Auch hier waren die Esterasen der Maus der humanen vergleichbar, die der Ratte waren wieder insensitiver.
Als Modell für die klinische Relevanz wurden HepG2-Zellen (eine humane Leberzellkarzinom-Zelllinie) und LS180-Zellen (eine humane Colonkarzinom-Zelllinie) mit dem Sapogenin Protopanoxadiol (PPD) behandelt, welches als Carbamat vorliegt und nach CES2-katalysierter Hydrolyse Zell-abtötende Aktivität entwickelt. Orlistat konnte die Effekte von PPD signifikant vermindern.
Schlussendlich wurde von den Autoren auch noch der Einfluss natürlich vorkommender genetischer Varianten in vitro getestet und teilweise Unterschiede in der CES2-Aktivität festgestellt.
CES2-Hemmung von klinischer Bedeutung?
Die Kenntnis, dass Orlistat CES2 inhibiert, ist neu. Die Frage stellt sich, ob diese in vitro beobachteten Effekte von klinischer Bedeutung sind. Hierzu müssten ausreichende Konzentrationen von Orlistat in der Leber erreicht werden. Nur in diesem Organ führt die Esterase-Aktivität zur Aktivierung von Prodrugs oder Eliminierung von Arzneistoffen. Wie oben angegeben, wurden in pharmakokinetischen Studien am Menschen Orlistat-Plasmakonzentrationen von bis zu 20 nmol/l gemessen. Diese gering erscheinenden Konzentrationen reichen der Studie von Xiao et al. zufolge aber aus. Bereits Konzentrationen von mehr als 1 nmol/l wiesen inhibitorische Aktivität auf und 50 nmol/l verursachten eine 50%ige CES2-Hemmung. Kritisch anzumerken ist allerdings, dass hierzu keine pharmakokinetischen Modelle angewendet wurden, also z. B. nicht mit unterschiedlichen Konzentrationen über verschiedene Zeiträume behandelt wurde, um zuverlässig die Inhibitionskonstanten zu ermitteln.
Irinotecan plus Orlistat: kaum vorstellbar!
Welche Arzneistoffe wären davon betroffen? Das in der Studie als Modell eingesetzte Irinotecan ist ein Topoisomerase-Hemmstoff u.a. zur Behandlung des Kolonkarzinoms. Es ist kaum vorstellbar, dass diese Patienten gleichzeitig mit einem peripheren Lipasehemmer behandelt würden, zu unsicher wäre die Bioverfügbarkeit anderer Arzneimittel, zu belastend wären die Nebenwirkungen.
Erhöhtes Blutungsrisiko bei Clopidogrel und Prasugrel?
Problematischer erscheinen die Thrombozytenaggregationshemmer Clopidogrel und Prasugrel. Clopidogrel wird zu 85% durch Esterasen inaktiviert [5], die Bioverfügbarkeit könnte dramatisch ansteigen, das Blutungsrisiko könnte sich erhöhen. Prasugrel hingegen wird in einer Zweischrittreaktion aktiviert, der erste Metabolisierungsschritt erfolgt vorwiegend durch CES2 [6]. Die Hemmung der Esterase würde somit zum Wirkungsverlust führen. Allerdings sind bislang keine Wechselwirkungen mit Orlistat – weder für Clopidogrel noch für Prasugrel – beschrieben worden. Es stellt sich auch die Frage, ob nicht die Thrombozytenaggregationshemmer durch die Steatorrhoe in ihrer Bioverfügbarkeit eingeschränkt werden könnten. Auch hier ist anzuraten, dass Patienten, die andere wichtige Arzneimittel einnehmen, nicht gleichzeitig mit Orlistat behandelt werden sollten.
Schlussendlich fehlt bislang der klinische Nachweis möglicher Interaktionen. Dies könnte nur durch gezielte klinische Studien erfolgen, bei der z. B. die Pharmakokinetik und Pharmakodynamik von Clopidogrel mit und ohne Orlistat an gesunden Probanden geprüft würden.
Fazit
Orlistat ist ein Arzneimittel, dessen langfristiger klinischer Nutzen unklar ist und welches durch seine Wirkung fettige Stühle bedingt, die zu einer Verminderung der Bioverfügbarkeit lipophiler Arzneistoffe führen können. Die in vitro beobachtete Esterasehemmung ist bislang nicht in vivo nachgewiesen worden. Allerdings sollten die Ergebnisse der Studie von Xiao et al. [1] zum Anlass genommen werden, Patienten, die Orlistat auf Rezept und auch ohne Rezept einnehmen, verstärkt nach der Einnahme anderer Arzneistoffe zu fragen. Da bislang keine Auffälligkeiten zum Wirkungsverlust z. B. von Clopidogrel bekannt wurden, besteht aber zurzeit noch kein Anlass zum akuten Eingreifen. Allerdings sollten die neuen Erkenntnisse zu möglichen Interaktionen auch dazu beitragen, über die Sinnhaftigkeit von Orlistat für die Gewichtsreduktion zu diskutieren.
Quelle[1] Xiao D, Shi D, Yang D, Barthel B, Koch TH, Yan B. Carboxylesterase-2 is a highly sensitive target of the antiobesity agent orlistat with profound implications in the activation of anticancer prodrugs. Biochem Pharmacol 2012.[2] Rucker D, Padwal R, Li SK, Curioni C, Lau DC. Long term pharmacotherapy for obesity and overweight: updated meta-analysis. BMJ 2007; 335 (7631): 1194 – 1199.[3] Padwal R, Li SK, Lau DC. Long-term pharmacotherapy for obesity and overweight. Cochrane Database Syst Rev 2004;(3):CD004094.[4] European databank on suspected adverse drug reaction reports. www.adrreports.eu/. 2013. [5] Trenk D, Kristensen SD, Hochholzer W, Neumann FJ. High on-treatment platelet reactivity and P2Y12 antagonists in clinical trials. Thromb Haemost 2012; 109 (2).[6] Williams ET, Jones KO, Ponsler GD, Lowery SM, Perkins EJ, Wrighton SA et al. The biotransformation of prasugrel, a new thienopyridine prodrug, by the human carboxylesterases 1 and 2. Drug Metab Dispos 2008; 36 (7): 1227 – 1232.
Autor
Prof. Dr. Dr. Ingolf Cascorbi, Institut für Experimentelle und Klinische Pharmakologie, Universitätsklinikum Schleswig-Holstein, Haus 30, Arnold-Heller-Str. 3, 24105 Kiel
Zum WeiterlesenLeberschäden unter OrlistatDas Antiadipositum Orlistat (Xenical®, 120 mg) wurde in Deutschland 1998 zugelassen. Seit 2009 ist auch ein nicht-verschreibungspflichtiges Präparat (Alli®, 60 mg) im Handel, inzwischen bieten auch Ratiopharm und Hexal Orlistat-Präparate an. Nachdem mehr als 20 Verdachtsfälle einer Leberschädigung nach der Einnahme von Orlistat bekannt geworden waren, wurde 2011 eine entsprechende Überprüfung eingeleitet. 2012 hat die EMA Orlistat zwar ein positives Nutzen-Risiko-Verhältnis bestätigt, jedoch veranlasst, dass die Fachinformationen auf die Möglichkeit sehr seltener Leberschädigungen hinweisen. |
DAZ 2013, Nr. 1/2, S. 32



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.