














- DAZ.online
- DAZ / AZ
- DAZ 48/2022
- Remdesivir, quo vadis

Foto: matho/AdobeStock
COVID-19
Remdesivir, quo vadis?
Der Einsatz des Nukleosid-Analogons im Wandel der Zeit
Lernziele
In diesem Beitrag lernen Sie unter anderem,
- wie Nukleosid-Analoga wirken,
- gegen welche Virusvarianten Remdesivir aktiv ist,
- für welche Patienten Remdesivir geeignet ist,
- in welcher Dosierung und wie lange die Behandlung empfohlen wird und
- auf welche Nebenwirkungen geachtet werden muss.
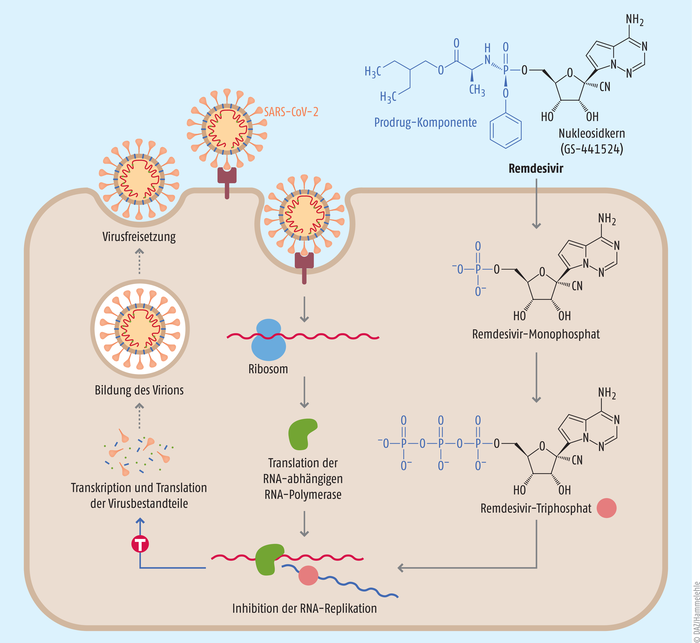
Der Wirkstoff Remdesivir (Veklury®) befand sich zum Ausbruch der COVID-19-Pandemie bereits länger in den Schubladen des Herstellers Gilead. Im Jahr 2009 wurde der Nukleosid-Precursor des Arzneistoffes, GS-441524 (s. Abb.), als möglicher Wirkstoff gegen Hepatitis C entwickelt [1]. Zwar wirkte das Compound nur schwach, stellte sich aber später als Breitband-Inhibitor von RNA-Viren heraus. Daraus entwickelten die Wissenschaftler des Konzerns 2013 das Monophosphoramidat-Prodrug Remdesivir, dessen antivirale Aktivität durch eine bessere Aufnahme in die Zellen erheblich verbessert wurde. In vitro wirkte der Arzneistoff z. B. gegen respiratorische Synzytial-Viren, Ebola-Viren, aber auch die gegen die SARS-CoV-2-Vorläufer SARS-CoV-1 und MERS-CoV und weitere (s. Tab. 1) [1]. Seine breite virustatische Wirkung entfaltet das Nukleosid-Analogon als Inhibitor der RNA-abhängigen RNA-Polymerase, eines Enzyms, das für die Replikation des Virusgenoms von RNA-Viren in der Wirtszelle zuständig ist (s. Abb.) [1]. Remdesivir wird von den Zellen aufgenommen und, dort angekommen, zum aktiven Triphosphat verstoffwechselt. Remdesivir-Triphosphat ähnelt dem natürlichen ATP und konkurriert mit dem Nukleotid um die RNA-abhängige RNA-Polymerase, welche den Wirkstoff in den entstehenden RNA-Strang einbaut. Danach kann das Enzym zwar noch ein paar Nukleotide anfügen, bis der Remdesivir-Baustein die Polymerase sterisch hemmt und einen Kettenabbruch induziert.
Abb.: Wirkweise des Nukleosid-Analogons Remdesivir Remdesivir greift als Inhibitor der RNA-abhängigen RNA-Polymerase des SARS-CoV-2 in die Replikation des Virus ein. Die Polymerase selbst wird durch die zelluläre Translationsmaschinerie der Wirtszelle ausgehend von der viralen RNA produziert. Das Phosphoramidat-Prodrug Remdesivir wird in die Zelle aufgenommen, zum Monophosphat hydrolysiert und schließlich durch Kinasen zum aktiven Triphosphat phosphoryliert. Durch seine Ähnlichkeit mit Adenosintriphosphat baut die RNA-abhängige RNA-Polymerase Remdesivir-Triphosphat in den RNA-Strang ein. Remdesivir hemmt dort nach dem Einbau weniger weiterer Nukleotide sterisch die Elongation des RNA-Strangs durch die Polymerase. Es kommt zum Kettenabbruch. Durch die gehemmte RNA-Replikation können weniger Virusbestandteile synthetisiert und Viren freigesetzt werden (vereinfacht nach [17]).
Auf der Schnellstraße zur Zulassung
Bereits während eines Ebola-Ausbruchs im Kongo 2018/2019 wurde der Wirkstoff im Rahmen einer klinischen Studie an Erkrankten untersucht [2]. Zwar unterlag Remdesivir in puncto Wirksamkeit den parallel untersuchten therapeutischen Antikörpern, aber zumindest konnte die Verträglichkeit des Wirkstoffes gezeigt werden. Nach Ausbruch der COVID-19-Pandemie wurde schnell gehandelt. Bereits Ende Januar 2020 wurde Remdesivir dem ersten nachgewiesenen COVID-19-Patienten in den USA im Rahmen eines Compassionate Use verabreicht, nachdem sich dessen Zustand verschlechterte [3]. Der Patient erholte sich schnell, und gleichzeitig fiel damit der Startschuss für das globale Compassionate-Use-Programm des Konzerns, das den Wirkstoff besonders schwer Erkrankten zugänglich machen sollte. Auch in Laborkulturen erwies sich Remdesivir als potenter Inhibitor des neuen Virus [4]. Remdesivir avancierte in dieser Zeit schnell zum Hoffnungsträger. Eine erste klinische Studie in China im Februar 2020 zeigte jedoch keinen Effekt des Arzneistoffes auf die Mortalität oder Zeit bis zur Genesung, musste aber auch vorzeitig abgebrochen werden, da die Zahl der Neuinfektionen in der Provinz stark einbrachen [5]. In Kooperation mit dem National Institute of Allergy and Infectious Diseases startete Gilead im gleichen Monat die internationale ACTT-1-Studie [6]. 1062 Probanden erhielten entweder Placebo oder Remdesivir (Loading Dose 200 mg i. v., gefolgt von 100 mg an den nächsten neun Tagen). Das Nukleosid-Analogon verkürzte die Genesungszeit signifikant von 15 Tagen unter Placebo auf nur noch zehn Tage (p < 0,0001). Die Mortalität verringerte sich zwar zahlenmäßig, aber am Ende der Behandlungszeit ohne statistische Signifikanz. Zwei weitere Open-Label-Studien zeigten, dass eine fünftägige Behandlung der zehntägigen ebenbürtig war, sodass potenziell mehr Patienten von der zu diesem Zeitpunkt nur begrenzt verfügbaren Substanz profitieren können [7, 8]. Die Interimsdaten dieser Studien genügten der FDA, um im Mai 2020 eine Notfallzulassung für Remdesivir zu erteilen, zunächst nur für hospitalisierte, schwer erkrankte Patienten, später für alle hospitalisierten Patienten. Die EMA folgte dem Beispiel und gab Remdesivir im Juli 2020 eine bedingte Zulassung für COVID-19-Patienten mit Pneumonie und Sauerstoffbedarf ab zwölf Jahren. Im Oktober 2020, kurz bevor die FDA das Therapeutikum regulär zuließ, dann der Paukenschlag: Die WHO verkündete erste Ergebnisse der Solidarity-Studie, die verschiedene potenzielle COVID-19-Therapeutika an über 14.000 Probanden systematisch untersucht hat. Laut der Interimsanalyse beeinflusste Remdesivir weder die Mortalität noch die Genesungszeit der Probanden [9]. In der späteren Auswertung der gesamten Studiendaten änderte sich die Grundtendenz zwar nicht, aber aufgrund der verbesserten statistischen Aussagekraft zeigte sich ein gewisser Nutzen: Remdesivir verminderte zumindest das Sterblichkeitsrisiko in der sauerstoffpflichtigen, nicht beatmeten Subgruppe um 13% gegenüber der Standardversorgung (Hazard Ratio 0,87; p = 0,03) [10]. In der aktuellen S3-Therapieleitlinie „Empfehlungen zur stationären Therapie von Patienten mit COVID-19“ wird aufgrund der heterogenen Studienlage keine Empfehlung für oder gegen das Präparat für hospitalisierte Patienten ausgesprochen [11]. Die WHO spricht mittlerweile eine bedingte Empfehlung (Conditional Recommandation) aus [12]. Sicher ist aber, dass Patienten, die bereits mechanisch beatmet werden, eine Remdesivir-Infusion nicht hilft. Von einem Einsatz bei diesem Patientenkollektiv rät die Leitliniengruppe ab.
Virusfamilie | Virus | mittlere effektive Konzentration (EC50) [µM] |
|---|---|---|
Filoviridae | Ebola-Virus (Ausbruch in Kikwit) | 0,19 |
Marburg-Virus | 0,06 | |
Coronaviridae | MERS-CoV | 0,07 |
SARS-CoV-1 | 0,07 | |
SARS-CoV-2 | 0,01 bis 1,6 | |
Pneumoviridae | humanes respiratorisches Synzytial-Virus | 0,015 |
Paramyxoviridae | Masern-Virus | 0,04 |
humanes Parainfluenza-Virus | 0,02 | |
Flaviviridae | Dengue-Virus | 0,25 |
Gelbfieber-Virus | 0,13 | |
Zika-Virus | 0,10 | |
West-Nil-Virus | 1,0 | |
Arenaviridae | Lassa-Virus | 4,5 |
Wende hin zur frühzeitigen Einnahme
Die Remdesivir-Story findet hier aber nur ihr vorläufiges Ende. Nach dem Ausbruch der Pandemie wurde der Wirkstoff dem Gebot der Stunde folgend vor allem an schwer erkrankten Patienten getestet. Gleichzeitig diskutierte die Fachwelt über eine Therapie im Frühstadium, während die Virusreplikation am höchsten ist, um zu verhindern, dass die Krankheit eskaliert [13]. Auch andere antivirale Wirkstoffe wie beispielsweise der Neuraminidase-Hemmer Oseltamivir (Tamiflu®) müssen generell frühzeitig gegeben werden. Gilead startete im September 2020 deshalb die PINETREE-Studie, die eine solche frühzeitige, dreitägige Remdesivir-Gabe an (ungeimpfte) Risikopatienten binnen sieben Tage nach Symptombeginn einer COVID-19-
Erkrankung untersuchte [14]. Gegenüber Placebo senkte das Nukleosid-Analogon den kombinierten Endpunkt aus Hospitalisierung und Tod um 87% (p < 0,0001). Die S3-Leitlinie zur stationären Therapie empfiehlt deshalb mittlerweile Remdesivir neben den Virustatika Nirmatrelvir/Ritonavir (Paxlovid®) und Molnupiravir (Lagevrio®) als Therapieoption für Risikopatienten, bis zu sieben Tage nach Infektionsbeginn [11]. Auch wenn die PINETREE-Studie an Ungeimpften durchgeführt wurde, gehen die Leitlinienautoren davon aus, dass analog zu anderen Virustatika auch für Geimpfte noch ein Benefit zu erwarten ist. In der Praxis wird Ritonavir-geboostetem Nirmatrelvir (Paxlovid®) gegenüber Remdesivir bei diesen Patientenkollektiven aber meist der Vorrang eingeräumt, da es als oral verfügbare Tablette einfacher verabreicht werden kann als das Nukleosid-Analogon, das an drei aufeinanderfolgenden Tagen infundiert werden muss. Jedoch interagiert der Ritonavir-Booster als CYP3A4-Inhibitor mit dem Metabolismus zahlreicher Arzneistoffe, sodass Remdesivir unter Umständen als wechselwirkungsärmere Alternative eingesetzt werden kann. Gilead arbeitet zudem an einem oral verfügbaren Prodrug des Wirkstoffes. Das gesamte Spektrum an Wechselwirkungen ist allerdings auch für Remdesivir zu diesem Zeitpunkt unbekannt. Aus In-vitro-Versuchen weiß man aber, dass Remdesivir von Esterasen in Gewebe und Plasma, und von den Cytochrom-Oxidoreduktasen CYP2C8, CYP2D6 und CYP3A4 verstoffwechselt wird, sowie Substrat für den organischen Anion-Transporter OATP1B1 und P-Glykoprotein ist [15]. Remdesivir selbst hemmt in vitro CYP3A4 sowie die organischen Anion-Transporter OATP1B1 und OATP1B3 und induziert CYP1A2 sowie potenziell CYP3A [15]. Inwieweit diese Interaktionen im klinischen Alltag zum Tragen kommen, ist derzeit nicht bekannt. Die Fachinformation empfiehlt deshalb, Patienten eng zu überwachen. Eine gleichzeitige Administration mit Dexamethason oder Baricitinib (Olumiant®) ist möglich.
Gute Verträglichkeit
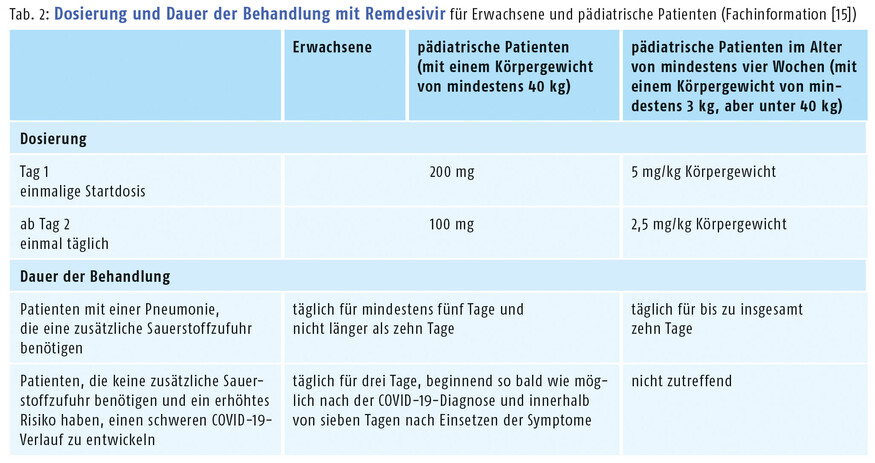
Erste Kenntnisse zur Sicherheit und Verträglichkeit des Medikaments bestehen hingegen schon seit der Ebola-Studie aus dem Jahr 2019. Die Zulassungsstudien stellen die Erkenntnisse mittlerweile auf eine breitere Basis. Zu den häufigsten Nebenwirkungen der Remdesivir-Infusionen zählen Kopfschmerzen, Übelkeit, Hautausschläge, erhöhte Transaminasen und eine verlängerte Prothrombinzeit [15]. Aufgrund einer in seltenen Fällen möglichen Anaphylaxie sollten die Patienten während der Infusion überwacht werden. Nicht zuletzt die gute Verträglichkeit führte dazu, dass die Zulassung von der EMA im September 2022 dann auch auf pädiatrische sauerstoffpflichtige bzw. nicht invasiv beatmete Patienten mit COVID-19-Pneumonie ab einem Alter von 28 Tagen (mindestens 3 kg Körpergewicht) ausgeweitet wurde (s. Tab. 2). Außerdem kann der Wirkstoff nun für nicht hospitalisierte pädiatrische Risikopatienten mit einem Mindestgewicht von 40 kg verordnet werden. In diesem Punkt unterscheidet sich Remdesivir von den beiden anderen Virustatika für Risikopatienten, Nirmatrelvir und Molnupiravir, die erst ab 18 Jahren eingesetzt werden können.
Omikron-Daten komplettieren Zulassung
Eine bedingte Zulassung, wie sie die Europäische Kommission im Juli 2020 für Veklury® ausgesprochen hat, bringt für den pharmazeutischen Hersteller auch die Pflicht mit sich, weitere Daten einzureichen. Ein vollständiger virologischer Bericht, der sich mit dem Resistenzprofil und der Wirksamkeit des Virustatikums z. B. auch gegenüber den aktuell zirkulierenden Virusvarianten auseinandersetzt, bildete die letzte Wissenslücke, die Gilead noch füllen musste. Der Hersteller konnte bestätigen, dass die Wirksamkeit gegenüber allen bisher aufgetretenen Varianten des Virus nicht abgenommen hat. Als nicht strukturgebendes Protein ist die RNA-abhängige RNA-Polymerase im Vergleich zum Spike-Protein wenig anfälliger für Mutationen. Bisher sind nur zwei Mutationen des Enzyms (P323L und G671S) in den verschiedenen Virusvarianten mit einer Prävalenz von mehr als 15% aufgetreten, die die Wirksamkeit aber nicht beeinträchtigten [16]. Aus In-vitro-Versuchen, bei denen das Virus in Gegenwart von Remdesivir subkultiviert wurde, weiß man aber, dass durchaus resistente Mutanten entstehen können. Ein kontinuierliches Monitoring bleibt daher unerlässlich. Mit dem Einreichen dieser letzten virologischen Daten konnte die bedingte Zulassung des Präparats in Europa schließlich in eine reguläre überführt werden. |
Interessenkonflikte
Der Autor versichert, dass keine Interessenkonflikte bestehen.
Literatur
[1] Cihlar T, Mackman RL. Journey of remdesivir from the inhibition of hepatitis C virus to the treatment of COVID-19. Antivir Ther 2022;27:13596535221082773
[2] Mulangu S et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N Engl J Med 2019;381:2293-2303
[3] Holshue ML et al. First Case of 2019 Novel Coronavirus in the United States. N Engl J Med 2020;382:929-936
[4] Pruijssers AJ et al. Remdesivir inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2-RNA polymerase in mice. Cell Rep 2020;32:107940
[5] Wang Y et al. Remdesivir in adults with severe COVID-19: a randomized, double-blind, placebo-controlled, multicentre trial. Lancet 2020;395:1569-1578
[6] Beigel JH et al. Remdesivir for the treatment of Covid-19 – final report. N Engl J Med 2020;383:1813–1826
[7] Goldman JD et al. Remdesivir for 5 or 10 days in patients with severe Covid-19. N Engl J Med 2020;383:1827–1837
[8] Spinner CD et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA 2020;324:1048–1057
[9] Solidarity Therapeutics Trial produces conclusive evidence on the effectiveness of repurposed drugs for COVID-19 in record time. Pressemitteilung WHO vom 15.10.2020. https://www.who.int/news/item/15-10-2020-solidarity-therapeutics-trial-produces-conclusive-evidence-on-the-effectiveness-of-repurposed-drugs-for-covid-19-in-record-time
[10] WHO Solidarity Trial Consortium. Remdesivir and three other drugs for hospitalised patients with COVID-19: final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet 2022;399:1941-1953
[11] Fichtner F et al. Empfehlungen zur stationären Therapie von Patienten mit COVID-19. S3-Leitlinie, Hrsg. Deutsche Gesellschaft für Internistische Intensivmedizin und Notfallmedizin (DGIIN), Deutsche Interdisziplinäre Vereinigung für Intensiv- und Notfallmedizin (DIVI), Deutsche Gesellschaft für Pneumologie und Beatmungsmedizin (DGP) und Deutsche Gesellschaft für Infektiologie (DGI). AWMF-Registernummer: 113-001LG, Stand September 2022, www.awmf.org/leitlinien
[12] World Health Organization. A living WHO guideline on drugs for Covid-19. BMJ 2020;370:m3379
[13] Siddiqi HK und Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J Heart Lung Transplant 2020;39:405-407
[14] Gottlieb RL et al. Early Remdesivir to Prevent Progression to Severe Covid-19 in Outpatients. N Engl J Med 2022;386:305-315
[15] Fachinformation Veklury® 100 mg Pulver für ein Konzentrat zur Herstellung einer Infusionslösung, Stand: September 2022
[16] Pitts J et al. Remdesivir and GS-441524 Retain Antiviral Activity against Delta, Omicron, and Other Emergent SARS-CoV-2 Variants. Antimicrob Agents Chemother 2022;66:e0022222
[17] Malin JJ et al. Remdesivir against COVID-19 and Other Viral Diseases. Clin Microbiol Rev 2020;34:e00162-20
Autor
Dr. Tony Daubitz, Studium der Pharmazie an der Universität Leipzig; Diplomarbeit in Basel an der Hochschule für Life Sciences der Fachhochschule Nordwestschweiz (FHNW) zu antientzündlichen Eigenschaften von Bambus-Extrakten; Promotion am Max-Delbrück-Centrum für Molekulare Medizin in Berlin zur Pharmakologie von Anionenkanälen



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.