
















- DAZ.online
- DAZ / AZ
- DAZ 43/2022
- Den „Eisen-Tod“ ...

Foto: Nomad_Soul/AdobeStock
Forschung
Den „Eisen-Tod“ verhindern
Bedeutung der antioxidativen Vitamine und der Vitamin-K-Familie
Über einen langen Zeitraum hinweg wurden die programmierte Apoptose (altgriechisch apóptosis, was im Deutschen „abfallen“ bedeutet, wie das Laub im Herbst von den Bäumen) sowie die unregulierte Nekrose als die beiden zentralen Zelltodmechanismen in Säugetieren erachtet. Mit dem Beginn des 21. Jahrhunderts kristallisierte sich jedoch heraus, dass eine Vielzahl ehemalig nekrotischer Prozesse im Grunde über definierte Zelltodsignalwege ablaufen. Diese umfassen Pyroptose, Nekroptose und eben auch die sogenannte Ferroptose (abgeleitet von dem lateinischen Wort ferrum für Eisen). Bei der Ferroptose handelt es sich um eine Eisen-abhängige Form des nicht apoptotischen Zelltods, die durch die exzessive Bildung von bestimmten reaktiven Sauerstoffradikalen (Reactive Oxygen Species, ROS) charakterisiert ist [1]. Während physiologische Konzentrationen an ROS für viele zelluläre Prozesse essenziell sind, führt eine überschießende Produktion dieser Sauerstoffradikale zu Membranschäden (oxidative Degradation von Lipiden, Lipidperoxidation) und konsekutivem Zelltod.
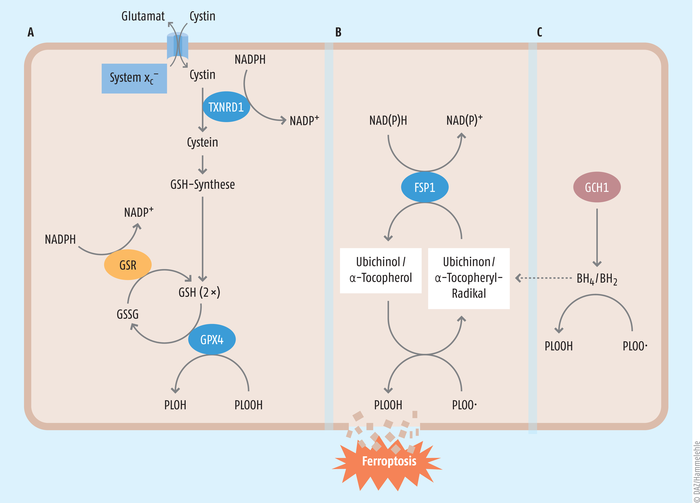
Obwohl die Namensgebung der Ferroptose im Jahr 2022 ihr erst zehnjähriges Jubiläum feiert, wird die wissenschaftliche Pionierarbeit bis in die 1950er-Jahre zurückdatiert [2]. Mit zunehmendem Verständnis der molekularen Prozesse, die entweder vor Ferroptose schützen oder diese begünstigen, stellte sich mittlerweile heraus, dass es sich um keine unkontrollierte zelluläre Produktion von reaktiven Sauerstoffradikalen handelt. Vielmehr stellen die Entstehung von Peroxiden in zellulären Membranbestandteilen (das heißt Phospholipidhydroperoxide) und speziell die Bildung von oxidierten Phosphatidylethanolaminen (PE) und Phosphatidylcholinen (PC) ein zentrales Ereignis der Ferroptose dar [3]. Phosphatidylethanolamine und Phosphatidylcholine zählen zu den Phospholipiden, die neben den Glykolipiden und Cholesterin zu den essenziellen Lipiden in biologischen Zellmembranen gehören und dadurch das Leben in seiner heutigen Form erst ermöglichen. Um einer unkontrollierten Lipidperoxidation und dem damit verbundenen ferroptotischen Zelltod entgegenzuwirken, haben sich auf molekularer Ebene drei Grundpfeiler antioxidativ wirkender Schutzmechanismen etabliert [4] (s. Abb. 1).
Abb. 1: Die drei bedeutendsten antiferroptotischen Schutzmechanismen Unter Verbrauch von Glutathion (GSH) werden Phospholipidhydroperoxide (PLOOH) zur ihren korrespondierenden Alkoholen (PLOH) über die Cystein/GSH/GPX4-Achse detoxifiziert (A). Dabei entstehendes Glutathiondisulfid (GSSG) wird mithilfe von NADPH wieder zu Glutathion recycelt. Anders als die Cystein/GSH/GPX4-Achse reduzieren die FSP1/Ubichinol-Achse (B) oder aber die GCH/BH4-Achse (C) das extrem gefährliche Phospholipidhydroperoxyl-Radikal zu einem Phospholipidhydroperoxid, wodurch eine Kettenreaktion, die sogenannte Lipidperoxidations-Kettenreaktion, und damit Ferroptose verhindert wird. GPX4: Glutathionperoxidase 4, FSP1: Ferroptosis-Suppressor-Protein 1, GSR: Glutathionreduktase, TXNRD1: Thioredoxinreduktase 1; BH2: Dihydrobiopterin; BH4: Tetrahydrobiopterin; GCH1: Guanosintriphosphat-Cyclohydrolase
- Der erste elementare Stützpfeiler ist die Selen-abhängige Glutathionperoxidase 4 (GPX4), da sie als einziges Redoxenzym im Säugerorganismus in der Lage ist, Phospholipid- und Cholesterolhydroperoxide unter Verbrauch von Glutathion direkt in den Zellmembranen zu reduzieren. Das Tripeptid Glutathion (GSH) ist das am häufigsten in Säugertierzellen vorkommende endogene Antioxidans und besteht aus den Aminosäuren Glycin, Glutamat und Cystein. Die Verfügbarkeit von Cystein stellt den limitierenden Faktor für die De-novo-Synthese von Glutathion in Zellen dar.
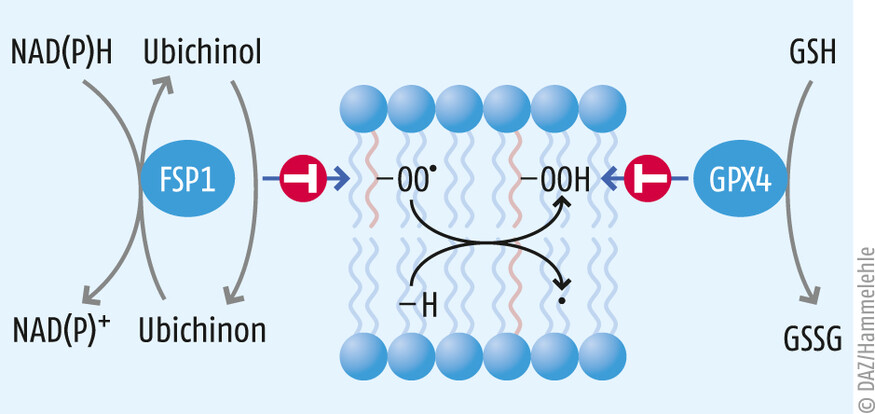
- Das zweite bedeutende antioxidative System ist das Ferroptose-Suppressor-Protein 1 (FSP1), welches extra-mitochondrial vorkommendes Ubichinon (Coenzym Q10, CoQ10) unter NAD(P)H-Verbrauch zu Ubichinol reduzieren kann [5]. Ubichinol seinerseits ist ein potentes lipophiles Antioxidans und vermag schädliche Lipidperoxidation und den damit verbundenen ferroptotischen Zelltod effektiv zu verhindern (s. Abb. 2).
- Das dritte antiferroptotisch wirksame System ist die Guanosintriphosphat-Cyclohydrolase (GCH1), als ratenlimitierendes Enzym der Synthese von Tetrahydrobiopterin (BH4) beschrieben [6]. BH4 ist ebenfalls ein wirksames lipophiles Antioxidans und wird fortlaufend durch die Dihydrofolatreduktase regeneriert. Das meiste unseres bisherigen Wissens über die Ferroptose basiert vornehmlich auf genetischen Deletionsmodellen oder aber pharmakologischen Inhibitorstudien, wobei gegenwärtig kein verlässlicher Biomarker zur Detektion ferroptotischen Zelltods etabliert werden konnte.
Pathophysiologische Relevanz der Ferroptose
Mittlerweile wurden viele humanrelevante Pathologien in unterschiedlichen Organsystemen mit Ferroptose in Verbindung gebracht [4], wobei insbesondere neurodegenerative Erkrankungen, Ischämie-/Reperfusions-bedingte Organschädigungen (ischemia/reperfusion injury, IRI) sowie verschiedene Tumorentitäten im Vordergrund stehen. Entsprechend ergeben sich hieraus auch unterschiedliche Therapieansätze oder zukünftige Behandlungsmöglichkeiten in Abhängigkeit davon, ob die gezielte Auslösung oder selektive Inhibierung von ferroptotischem Zelltod im Fokus steht.
Gezielte Ferroptose-Induktion
Der Begriff der Ferroptose wurde 2012 in Zusammenhang mit einer humanen Fibrosarkom-Zelllinie geprägt, die onkogenes Ras überexprimierte. Mittlerweile konnte in Zellkulturexperimenten jedoch vielfach nachgewiesen werden, dass ferroptotischer Zelltod in vielen unterschiedlichen Tumorzellreihen auch ohne onkogenes Ras ausgelöst werden kann, z. B. in klarzelligen Karzinomen der Nieren sowie Ovarien, im duktalen Pankreaskarzinom, im hepatozellulären Karzinom oder im diffus großzelligen B-Zell-Lymphom. Darüber hinaus zeigten sich dreifach-negative Brustkrebszelllinien (mangelnde Oberflächenexpression von Estrogen-/Progesteron-Rezeptoren oder des humanen epidermalen Wachstumsfaktor-Rezeptors HER2) sensitiv gegenüber einer selektiven Ferroptose-Auslösung [7]. Auch entwickeln insbesondere standardtherapieresistente und metastasierende Tumoren eine ausgesprochen hohe Vulnerabilität gegenüber Ferroptose [8]. Hierdurch könnte zukünftig eine therapeutische Nische im Sinne personalisierter Medizin für einen prognostisch ungünstigen und konventionell nur schwer zugänglichen Subtyp entstehen. Es gibt aber noch keine selektiven und hochwirksamen Ferroptose-Induktoren mit überzeugendem pharmakokinetischem und pharmakodynamischem Profil, um diese in präklinischen Mausmodellen zu prüfen.
Selektive Ferroptose-Inhibition
Dagegen konnte eine Eisen-abhängige Lipidperoxidation bereits bei Morbus Alzheimer und Morbus Parkinson und z. B. auch bei der amyotrophen Lateralsklerose oder bei Chorea Huntington nachgewiesen werden. Weiterhin spielt die Ferroptose eine bedeutende Rolle bei Ischämie-/Reperfusions-bedingten Organschädigungen des Gehirns nach einem ischämischen Schlaganfall und des Herzens infolge eines akuten Myokardinfarktes oder der Leber und Nieren. Gerade bei den letzteren Organen verlangt eine Verhinderung ferroptotischen Zelltods besondere Aufmerksamkeit aus dem Blickwinkel solider Organtransplantationen.
Anders als die gezielte Ferroptose-Induktion werden selektive Inhibitoren der Ferroptose bereits klinisch getestet. Mechanistisch steht hier die Eisenchelation oder die Verhinderung überschießender Lipidperoxidation im Vordergrund. Deferiprone (Ferriprox®) ist ein von der FDA in den USA zugelassener Eisenchelator zur Behandlung einer transfusionsassoziierten Eisenüberladung im Rahmen einer Thalassämie oder Sichelzellanämie. Darüber hinaus wird Deferiprone in den USA gegenwärtig zur Behandlung von Patienten mit M. Alzheimer (Phase II), M. Parkinson (Phase II) oder amyotropher Lateralsklerose (Phase II/III) in frühen Krankheitsstadien klinisch getestet. Zur Verhinderung schädlicher Lipidperoxidation kristallisierte sich in einem High-Throughput-Screening das Spiroquinoxalinamin-Derivat Liproxstatin 1 (Lip-1) als hochpotenter, lipophiler Radikalfänger und spezifischer Inhibitor der Ferroptose heraus. Liproxstatin 1 kann oxidierte Phospholipide in Liposomen und speziell in Biomembranen effektiv reduzieren [9]. Daneben besitzt Lip-1 ein vielversprechendes pharmakokinetisches/pharmakodynamisches Profil, und durch chemische Veränderungen des Grundmoleküls wurden Liproxstatine der nächsten Generation (Lip-2) entwickelt, die sich selektiv im Gewebe anreichern. Die Entwicklung von Lip-2 befindet sich bereits am Übergang in die klinische Studienphase, dabei soll die Toxizität im Menschen erstmalig im Rahmen einer postmortalen Lebertransplantation als Pilotprojekt geprüft werden.
Bedeutung antioxidativer Vitamine
Bereits in den 1950er-Jahren konnte der Zusammenhang zwischen der Menge an ungesättigten Fettsäuren, Eisen-Akkumulation und schädlicher Lipidperoxidation in Kleinnagern hergestellt werden, die durch Nahrungsergänzung von Vitamin E gemildert werden konnte [10].
Vitamin E wird als Nahrungsergänzungsmittel oder in der chemischen Industrie verwendet und ist ein Sammelbegriff von acht natürlich vorkommenden, lipophilen Antioxidanzien. Hierzu zählen α-, β-, γ- und δ-Tocopherol sowie α-, β-, γ- und δ-Tocotrienol, wobei α-Tocopherol die biologisch aktivste Substanz darstellt. Um die übermäßige Ausbildung von oxidierten Phospholipiden mit anschließend dominoeffektartiger Ausbreitung zu verhindern, bieten sich drei Herangehensweisen an:
- die Reduktion des Angebots oxidierbarer Phospholipide („Zunder”),
- die Verhinderung der Radikalinitiation („Zündfunke”) und
- der anschließend exponentiellen Ausbreitung („Lauffeuer” = Lipidperoxidation).
Als lipophiler Radikalfänger unterdrückt α-Tocopherol diese Kettenreaktion, indem es mit gebildeten Peroxylradikalen (diese sind die eigentlichen Treiber der Lipidperoxidationskettenreaktion) in der Phospholipid-Doppelschicht reagiert und diese entschärft. Obwohl sich α-Tocopherol in Zellkultur als wirksamer Hemmstoff der Ferroptose herausstellte [11], müssen Tierversuche differenziert betrachtet werden, da in kommerziell verfügbarem Tierfutter häufig supraphysiologische Konzentrationen von Vitamin E enthalten sind.
Der biochemisch relevante Unterschied zu einem nicht natürlich vorkommenden Ferroptose-Inhibitor wie z. B. Lip-1 und Lip-2 besteht primär in der funktionellen Gruppe. Während α-Tocopherol eine Chromanolgruppe als reaktive Seitengruppe aufweist, enthält Lip-1 eine funktionelle Aminogruppe. Hierdurch hat α-Tocopherol zwar hervorragende antioxidative Eigenschaften in organischen Lösungsmitteln, die jedoch nicht unreflektiert auf Biomembranen übertragbar sind. Insbesondere in Lysosomen ist Lip-1 ein wesentlich potenterer Ferroptose-Inhibitor [12].
Bei der Ascorbinsäure handelt es sich um den vermutlich prominentesten Vertreter der Vitamine. Im menschlichen Körper nimmt Vitamin C zahlreiche Funktionen wahr und kann mitunter auch oxidiertes α-Tocopherol regenerieren. Eine überzeugende antiferroptotische Wirkung im Menschen ließ sich allerdings bislang nicht nachweisen.
Bedeutung der Vitamin-K-Familie
Vitamere der Vitamin-K-Familie konnten durch systematische Suche nach natürlich vorkommenden Inhibitoren der Ferroptose identifiziert werden 13]. Neben Phyllochinon (Vitamin K1) war insbesondere auch hochdosiertes Menachinon 4 (MK-4; Vitamin K2) in der Lage, vor ferroptotischem Zelltod sowohl in Zellkultur als auch im Rahmen eines Mausmodells der Leber und Nieren zu schützen. Physiologisch kommt Phyllochinon in Pflanzen vor und wird nach Aufnahme über den Verdauungstrakt gewebeabhängig zu MK-4 umgewandelt. Neben MK-4 kommen auch Vitamin-K2-Vertreter mit einer längeren Seitengruppe (Isoprenoid-Seitenketten, z. B. MK-7, MK-9) vor, die hauptsächlich von fakultativ (bzw. obligat) anaeroben Bakterien synthetisiert werden. Die größte natürlich vorkommende Quelle an Vitamin K2 sind durch die Einwirkung von Bacillus subtilis fermentierte Sojabohnen, auch als japanisches Nattō bekannt. Bei Menadion (Vitamin K3) handelt es sich um ein synthetisches Molekül mit ebenfalls antiferroptotischer Wirksamkeit.
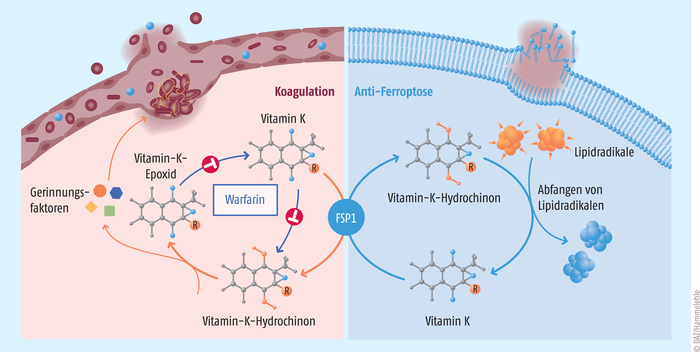
Klinisch ist Vitamin K essenzieller Kofaktor für die Blutgerinnung und den Knochenmetabolismus. Diese Entdeckung wurde bereits 1943 mit dem Nobelpreis für Physiologie oder Medizin prämiert. Vollständig reduziertes Vitamin K (Vitamin-K-Hydrochinon, VKH2) dient dabei als Kofaktor der γ-Glutamylcarboxylase (GGCX) und wird zu einem Vitamin-K-2,3-Epoxid (VKO) umgewandelt (Abb. 3). In der Leber werden die Vitamin-K-abhängigen Gerinnungsfaktoren II, VII, IX und X durch eine γ-Glutamylcarboxylase-katalysierte Einführung einer Carboxygruppe in die γ-Position von bestimmten Glutamylresten synthetisiert. Das dabei entstehende Vitamin-K-2,3-Epoxid wird in zwei Schritten durch die Vitamin-K-Epoxid-Reduktase (VKORC) zu Vitamin-K-Hydrochinon regeneriert. Und genau dieses Enzym ist das Zielmolekül von Warfarin, eines der am häufigsten eingesetzten indirekten Vitamin-K-Antagonisten und Blutgerinnungshemmer. Neben der Warfarin-sensitiven Vitamin-K-Epoxid-Reduktase wurde seit mehr als einem halben Jahrhundert auch noch ein Warfarin-resistentes Protein postuliert, dessen Identität allerdings bis dato unbekannt war. In den von Mishima et al. [13] durchgeführten Studien wurde zunächst untersucht, ob eine vorbeugende MK-4-Gabe zell- und gewebeprotektive Effekte hat. Bestimmt wurde die Outcome-Verbesserung einer subletalen hepatischen und renalen Ischämie-/Reperfusions-bedingten Organschädigung. Verglichen mit den Kontrolltieren konnten histopathologisch in der Leber weniger nekrotische Areale und auch weniger Lipidperoxidation dokumentiert werden. Auch in den Nieren ließen sich mikroskopisch weniger ferroptotische Zellen nachweisen. In diesem Versuchsansatz bestätigte sich der antioxidative Effekt von MK-4 zur Reduktion schädlicher Lipidperoxidation und letztlich Ferroptose.
Abb. 3: Die zwei Funktionen von Ferroptosis-Suppressor-Protein 1 (FSP1) Einerseits kann FSP1 Vitamin K zu Vitamin-K-Hydrochinon reduzieren, um Phospholipidhydroperoxyl-Radikale abzufangen und die damit verbundene Ferroptose zu unterbinden (rechts). Andererseits kann durch die Reduktion von Vitamin K über das Ferroptosis-Suppressor-Protein 1 eine Warfarin-induzierte Hemmung der Vitamin-K-Epoxid-Reduktase umgangen und eine Normalisierung der Prothrombinzeit erreicht werden.
Darüber hinaus wurden Versuchstiere einer Überdosis Warfarin ausgesetzt und begleitend mit MK-4 behandelt. Das Besondere an den Versuchstieren war, dass diese entweder nur ein (Fsp1+/-) oder gar kein Fsp1-Allel (Fsp1-/-) enthielten. Nur bei den Fsp1+/--Mäusen wurden unter MK4-Behandlung eine normale Überlebensrate und Blutgerinnung trotz Warfarin-Überdosierung beobachtet. Hieraus lässt sich ableiten, dass MK-4 im Mausmodell auch unabhängig von der Vitamin-K-Epoxid-Reduktase über FSP1 reduziert werden kann und dadurch letale innere und insbesondere intrazerebrale Blutungen infolge einer Warfarin-Überdosierung verhindert wurden. Diese Befunde konnten durch enzymatische und zellkulturbasierte Experimente eindeutig belegt werden, sodass man schlussfolgern kann, dass es sich bei FSP1 um das lang postulierte Warfarin-resistente Enzym im bekannten Vitamin-K-Zyklus handeln muss.
Allerdings muss bei den In-vivo-Versuchen darauf hingewiesen werden, dass in den einzelnen Mausmodellen supraphysiologische Dosierungen von MK-4 eingesetzt wurden. Gewichtsadaptiert entspräche dies in Bezug auf eine Person mit einem Körpergewicht von 75 kg im Rahmen der Modelle zu Ischämie-/Reperfusions-bedingten Organschäden einer kurzzeitig verwendeten MK-4-Dosis von 15 g und in Bezug auf eine Warfarin-Überdosierung einer täglich parenteralen Zufuhr von 750 mg bis 1500 mg. Zum Vergleich enthalten handelsübliche Vitamin-K1-Ampullen 10 mg und Vitamin-K2-Tabletten 200 µg des Wirkstoffes. Die Analyse einer möglicherweise zunehmenden Lipidperoxidation durch Warfarin oder sogar Induktion ferroptotischen Zelltods war kein Teil der Versuchsanordnung, so dass darüber zum gegenwärtigen Zeitpunkt nur spekuliert werden kann. Insofern steht die indikationsgerechte Anwendung von Cumarin-Derivaten beispielsweise zur oralen Antikoagulation nach mechanischem Herzklappenersatz oder aber infolge eines thromboembolischen Ereignisses im Rahmen eines Antiphospholipid-Syndroms außer Frage.
Fazit
Unsere Forschung identifiziert Vitamin K als ein in der Natur vorkommendes potentes antiferroptotisches Agens. Überraschenderweise konnte darüber hinaus gezeigt werden, dass das Ferroptosis-Suppressor-Protein 1 die treibende Kraft hinter dieser antiferroptotischen Funktion von Vitamin K ist und einen ungewöhnlichen Vitamin-K-Zyklus aufrechterhält, welcher ebenfalls Zellen effizient vor Ferroptose schützen kann. Zudem konnten wir eindeutig belegen, dass FSP1 das Warfarin-resistente Enzym ist, das innerhalb des bekannten Vitamin-K-Zyklus zwar schon lange vermutet wurde, bisher jedoch unbekannt war. Hierbei sind Phyllochinon und Menachinon die essenziellen Elektronenüberträger bei photosynthetisch aktiven Pflanzen bzw. fakultativ anaeroben Bakterien, weshalb wir vermuten, dass das FSP1-Vitamin-K-System eine noch weitreichendere Bedeutung als „nur“ beim Säugerorganismus haben könnte. |
Literatur
[1] Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060-1072
[2] Eagle H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J Exp Med 1955;102:37-48
[3] Kagan VE et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81-90
[4] Zheng J, Conrad M. The Metabolic Underpinnings of Ferroptosis. Cell Metab 2020;32:920-937
[5] Doll S et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019;75:693-698
[6] Kraft VAN et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 2020;6:41-53
[7] Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017;13:91-98
[8] Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017;551:247-250
[9] Friedmann Angeli JP et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014;16:1180-1191
[10] Golberg L, Smith JP. Changes associated with the accumulation of excessive amounts of iron in certain organs of the rat. Br J Exp Pathol 1958;39:59-73
[11] Seiler A et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 2008;8:237-248
[12] Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol 2019;15:1137-1147
[13] Mishima E et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022;608:778-783
Autoren
Tobias Seibt, Studium der Humanmedizin an der Ludwig-Maximilians-Universität München (LMU); Facharzt für Innere Medizin und Nephrologie im Transplantationszentrum München der LMU sowie Physician Scientist am Helmholtz Zentrum München
Adam Wahida, Medizinstudium in Aachen (RWTH) und München (TUM); forscht als Arzt am Helmholtz Zentrum München sowie am Nationalen Centrum für Tumorerkrankungen (NCT) in Heidelberg
Eikan Mishima, Medizinstudium an der Tohoku Universität (Sendai, Japan); arbeitet als Physician Scientist am Helmholtz Zentrum München
Marcus Conrad, Biologiestudium an der Universität Konstanz, Promotion an der Ludwig-Maximilians-Universität München/Helmholtz Zentrum München; Leiter des Instituts für Metabolismus und Zelltod am Helmholtz Zentrum München



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.