













- DAZ.online
- DAZ / AZ
- DAZ 36/2022
- Krebsmedikament ...
Arzneimittel und Therapie
Krebsmedikament Amivantamab vom Markt genommen
Der bispezifische Tyrosinkinase-Inhibitor Amivantamab (Rybrevant®) wurde im Januar 2022 als Monotherapeutikum für Erwachsene mit fortgeschrittenem nicht-kleinzelligem Lungenkarzinom (non-small cell lung cancer, NSCLC) und aktivierender Exon-20-Insertionsmutation in Deutschland eingeführt. Da die Zulassung nur aufgrund einer nicht-randomisierten Studie erfolgt war, hat nun der Gemeinsame Bundesausschuss (G-BA) keinen Zusatznutzen gegenüber der zweckmäßigen Vergleichsmedikation anerkannt und damit eine Marktrücknahme ausgelöst. Trotz der Verdopplung der Überlebenszeit in der zulassungsrelevanten Studie gegenüber historischen Vergleichen bleibt die hochwirksame Substanz nun den betroffenen Patienten verwehrt.
Exon-20-Insertion als Treibermutation
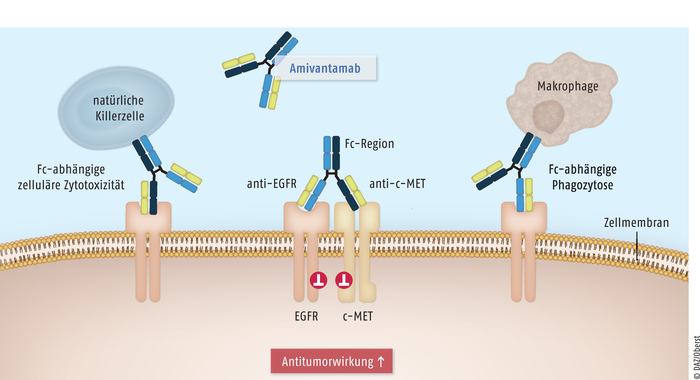
Lungenkarzinome gehören zu den malignen Erkrankungen mit der höchsten Zahl an genetischen Veränderungen. Bei Nachweis einer behandelbaren Treibermutation werden im Rahmen moderner Therapieregime zunehmend zielgerichtete Therapien eingesetzt. Exon-20-Insertionen stellen mit einem Anteil von 12% die dritthäufigsten aktivierenden Mutationen im Epidermalen-Wachstumsfaktor-Rezeptor(EGFR)-Gen dar. Die Betroffenen, bei denen es sich zu einem großen Teil um Nichtraucher handelt, haben im Vergleich zu NSCLC mit anderen EGFR-Mutationen eine ungünstigere Prognose. Meist besteht eine Resistenz gegen Tyrosinkinase-Inhibitoren der ersten, zweiten und dritten Generation, sodass bislang lediglich eine Platin-basierte Chemotherapie durchgeführt werden konnte. Nur etwa 8% der Patienten erreichten eine Überlebenszeit von fünf Jahren. Mit dem bispezifischen Antikörper Amivantamab ist nun erstmals ein zielgerichteter Wirkstoff für diese Patienten mit Exon-20-Insertionsmutation verfügbar. Der Wirkmechanismus mit kombinierter Hemmung der Signalwege von EGFR und der weiteren Tyrosinkinase mesenchymal-epithelialer Transitionsfaktor (c-MET) ist innovativ. Zusätzlich führt Amivantamab zu zellulärer Zytotoxizität sowie Phagozytose durch natürliche Killerzellen und Makrophagen. Als Folge kann bei den NSCLC-Patienten ein relevanter Gewinn an Überlebenszeit erreicht werden.
Der bispezifische Antikörper Amivantamab blockiert bei NSCLC-Tumorzellen mit aktivierender Mutation die Ligandenbindung an die Tyrosinkinasen epidermaler Wachstumsfaktor-Rezeptor (EGFR) und mesenchymal-epithelialer Transitionsfaktor (c-MET) und damit die nachgeschalteten tumorfördernden Signalkaskaden. Zur Antitumorwirkung tragen eine verstärkte EGFR- und c-MET-Degradierung und eine Fc-Domänen-vermittelte zelluläre Zytotoxizität und Phagozytose durch natürliche Killerzellen und Makrophagen bei.
Wöchentliche, später zweiwöchentliche Infusionen
Vor Beginn einer Amivantamab-Therapie muss ein positiver EGFR-Exon-20-Insertionsmutationsstatus nachgewiesen werden. Die Substanz wird bei Patienten unter 80 kg Körpergewicht in Einzeldosen von 1050 mg intravenös gegeben. Patienten ab 80 kg erhalten entsprechend 1400 mg pro Dosis. Die Applikation findet in den ersten vier Wochen wöchentlich, danach alle zwei Wochen statt. Die Infusionsgeschwindigkeit liegt bei Therapiebeginn im Bereich von 50 ml/h und kann bei den späteren Applikationen und guter Verträglichkeit auf bis zu 125 ml/h gesteigert werden. Bei schweren Nebenwirkungen sollte die Dosis reduziert oder die Applikation bis zum Eintritt einer Besserung unterbrochen werden, allerdings müssen nach Therapiepausen von mehr als sieben Tagen zunächst stets reduzierte Dosen zum Einsatz kommen. Patienten mit schwerer Nieren- oder mäßiger bis schwerer Leberinsuffizienz dürfen aufgrund fehlender Erfahrungen nur mit Vorsicht und unter Überwachung der Nebenwirkungen mit Amivantamab behandelt werden.
Wichtige Vorsichtsmaßnahmen
Infusionsbedingten Reaktionen sollte durch die Gabe einer Prämedikation aus Antihistaminika und Antipyretika begegnet werden. Vor allem bei der ersten Infusion empfehlen sich die zusätzliche Applikation von Glucocorticoiden und die Aufteilung der Dosis auf zwei aufeinanderfolgende Tage. Bei wiederholtem Auftreten von schweren Reaktionen ist ein dauerhaftes Absetzen von Amivantamab angeraten. Die Therapie mit dem Antikörper ist mit interstitiellen Lungenerkrankungen assoziiert. Auf entsprechende Symptome wie Dyspnoe, Husten oder Fieber ist zu achten. Bei bestätigter Diagnose muss die Amivantamab-Behandlung beendet werden. Falls es während der Therapie zu Haut- und Nagelreaktionen kommt, können je nach Schweregrad Pflegecremes sowie topische und systemische Corticosteroide und Antibiotika zum Einsatz kommen. Bei lebensbedrohlichen Hautreaktionen wie einer toxisch-epidermalen Nekrolyse ist die Behandlung mit dem Antikörper dauerhaft abzusetzen. Die Patienten müssen zudem während und bis zu zwei Monate nach der Beendigung der Amivantamab-Therapie die Sonne meiden und entsprechende Schutzkleidung tragen bzw. UV-A-/UV-B-Breitband-Sonnenschutzmittel anwenden. Während der Anwendung von Amivantamab muss mit Augenerkrankungen wie Keratitis gerechnet werden. Bei sich verschlechternder Augensymptomatik sollte das Tragen von Kontaktlinsen vermieden und umgehend ein Ophthalmologe zu Rate gezogen werden. Andere EGFR- und c-MET-Inhibitoren zeigten im Tierexperiment vermehrte embryofetale Entwicklungsstörungen und Sterblichkeit bzw. Aborte. Somit könnte auch Amivantamab zu einer Schädigung des ungeborenen Kindes führen. Die Anwendung während der Schwangerschaft darf daher allenfalls nach strenger Nutzen-Risiko-Abwägung stattfinden.
CHRYSALIS-Studie
Die Zulassung von Amivantamab beruht auf der offenen Phase-Ⅰ-Studie an 114 NSCLC-Patienten mit lokal fortgeschrittenem oder metastasiertem Tumor. In die Untersuchung wurden Erwachsene mit Exon-20-Insertionsmutation und Versagen einer vorangegangenen Platin-basierten Therapie aufgenommen. Die Responserate lag bei 40%, wobei drei Patienten ein vollständiges Ansprechen auf Amivantamab mit vollständigem Verschwinden der Tumorzeichen zeigten. Die mediane Responsedauer betrug 11,1 Monate, die mediane Überlebenszeit ohne Erkrankungsprogression wurde mit 8,3 Monaten angegeben. Bei 86% der Teilnehmer kam es während der Therapie zu Hautausschlägen, 66% erlitten infusionsbedingte Reaktionen (vor allem nach der ersten Applikation) und 45% Nagelbettentzündungen. Bei 13% der Patienten musste aufgrund von Behandlungs-assoziierten UAW eine Reduktion der Amivantamab-Dosis erfolgen, bei 4% war ein Therapieabbruch erforderlich.
Zusatznutzen nicht anerkannt
Zahlreiche Fachgesellschaften, beispielsweise die Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie, hatten sich im Vorfeld für die Festlegung eines Zusatznutzens durch Amivantamab ausgesprochen. Dem wurde im Juli 2022 durch den G-BA im Rahmen der frühen Nutzenbewertung neuer Arzneimittel widersprochen. Der Ausschuss bemängelte, dass die Zulassung lediglich aufgrund einer nicht-randomisierten Studie erfolgt war. Ein indirekter Vergleich mit Daten von weltweit geführten Patientenregistern zeigt zwar nahezu eine Verdopplung der medianen Überlebenszeit an, er wurde jedoch vom G-BA als nicht valide eingestuft. Ende August 2022 hat nun die Firma Janssen-Cilag bekannt gegeben, das hochwirksame Krebsmedikament mit sofortiger Wirkung vom deutschen Markt zu nehmen. Für die Patientinnen und Patienten mit prognostisch ungünstiger Exon-20-Insertionsmutation ist das sehr bedauerlich. Zur Fortsetzung einer bereits begonnenen Therapie oder zur Einleitung einer neuen Behandlung kann Amivantamab nun allenfalls mit hohem Aufwand aus dem Ausland importiert werden. |
Literatur
[1] Fachinformation zu Rybrevant®, Stand Dezember 2021
[2] Park K, Haura EB, Leighl NB, Mitchell P et al. Amivantamab in EGFR exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS Phase I Study. J Clin Oncol 2021;39(30):3391-3402
[3] Neubeck M, Mutschler E. Amivantamab. Neue Arzneimittel 2022;4:66-68
[4] Nutzenbewertungsverfahren zum Wirkstoff Amivantamab (Lungenkarzinom, nicht-kleinzelliges, EGFR-Exon-20-Insertionsmutation, nach platinbasierter Therapie). Gemeinsamer Bundesausschuss (G-BA), Beschlussfassung 7. Juli 2022



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.