













- DAZ.online
- DAZ / AZ
- DAZ 12/2022
- Wie „nützlich“ sind ...
Arzneimittel und Therapie
Wie „nützlich“ sind Orphan Drugs?
Auch Arzneimittel für seltene Krankheiten müssen ihre klinische Effektivität zeigen
Direktive 141/2000 zu Arzneimitteln für seltene Leiden definiert die Bedingungen, unter denen ein Wirkstoff als Orphan Drug anerkannt werden kann:
- Das Medikament soll zur Diagnose, Verhütung oder Behandlung eines Leidens, das lebensbedrohend ist oder eine chronische Invalidität nach sich zieht, dienen,
- zum Zeitpunkt der Antragstellung sind in der EU nicht mehr als fünf von 10.000 Personen betroffen (Prävalenz der Krankheit) und
- das Inverkehrbringen des Arzneimittels in der EU würde ohne Anreize vermutlich nicht genügend Gewinn bringen, um die notwendigen Investitionen zu rechtfertigen.
Des Weiteren gilt:
- Es wurde in der Gemeinschaft noch keine zufriedenstellende Methode für die Diagnose, Verhütung oder Behandlung des betreffenden Leidens zugelassen, oder
- das betreffende Arzneimittel wird – sofern eine solche Methode besteht – für diejenigen, die von diesem Leiden betroffen sind, von erheblichem Nutzen sein.
Die Anerkennung als Orphan Drug erfolgt zentral bei der European Medicines Agency (EMA) und zwar durch einen dafür speziell gebildeten Ausschuss (Committee for Orphan Medicinal Products, COMP). Dieser besteht aus Experten, Vertretern der nationalen Arzneimittelagenturen und Vertretern von Patientengruppen und muss über einen Antrag innerhalb von 90 Tagen entscheiden. Eine Anerkennung bedeutet nicht gleichzeitig die Zulassung eines Arzneimittels. Letztere erfolgt, wenn auch beschleunigt, ebenfalls zentral durch die auch für andere Medikamente gültige Zulassungsprozedur der EMA (s. Abb. 1).
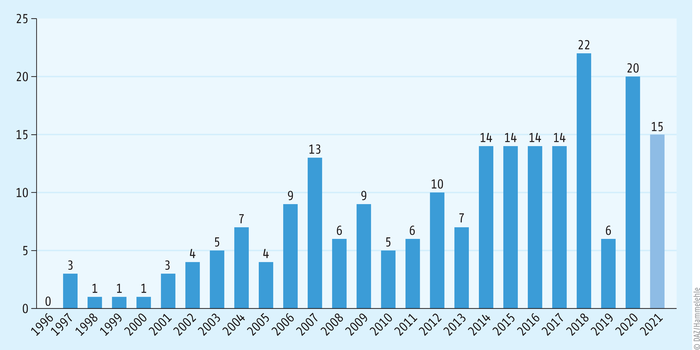
Abb. 1: Zahl der Orphan Drugs in der Europäischen Union Aktuell (Stand: Dezember 2021) werden 129 zugelassene Medikamente mit aktivem Orphan-Drug-Status gezählt; dazu kommen noch 68 Medikamente, die den Status früher einmal hatten (fast alle davon sind noch auf dem Markt). 2000 trat die europäische Orphan-Drug-Verordnung in Kraft (nach Informationen des vfa [5]).
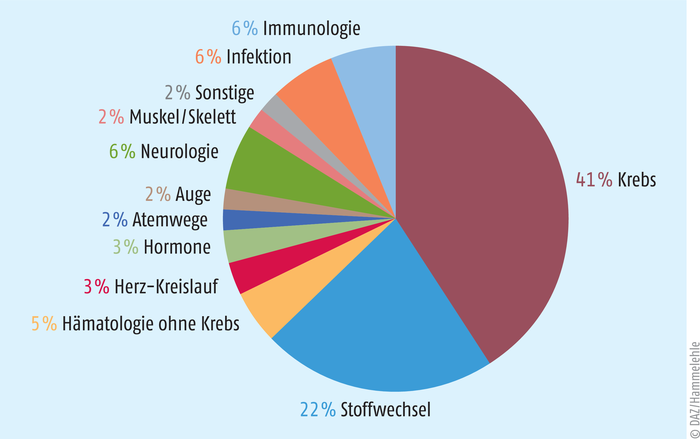
Eine aktuelle Liste der in Europa anerkannten und zugelassenen Orphan Drugs ist auf der Seite der EU zu finden [2]. Diese enthält 1856 Substanzen (Stand: Februar 2022) von denen allerdings nur 159 sich auch als zugelassene Arzneimittel auf dem europäischen Markt befinden. Davon sind wiederum 45 Produkte für die Behandlung von hämatologischen und onkologischen Krankheiten indiziert (s. Abb. 2).
Abb. 2: Anwendungsgebiete Insgesamt erhielten Arzneimittel gegen 152 Krankheiten eine Orphan-Drug-Zulassung (nach Informationen des vfa [5]).
Welche Vorteile hat die Anerkennung als Orphan Drug?
Für eine schnelle Zulassung sind gut geplante Studien notwendig, die auch von der EMA anerkannt werden. Für Orphan Drugs bietet die EMA eine spezifische Protokoll-Beratung zu reduzierten Gebühren, die für universitäre Einrichtungen kostenfrei ist. Ebenfalls reduziert, besonders für kleinere Firmen, sind die Zulassungsgebühren. Kleinere Firmen können dazu noch Fördermittel erhalten. Darüber hinaus genießen Orphan Drugs einen zusätzlichen Patentschutz von zehn Jahren, der bei pädiatrischen Indikationen (60% der Anträge) um weitere zwei Jahre verlängert wird. Die EMA arbeitet bei der Zulassung von Orphan Drugs auch intensiv mit der USA und der japanischen Behörden zusammen. Dadurch werden auch internationale Zulassungen erheblich vereinfacht. In Deutschland müssen sich Orphan Drugs mit einem Jahresumsatz von weniger als 50 Millionen Euro nach dem Marktzugang keinem regulären Nutzenbewertungsverfahren gemäß §35a SGB V durch das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) unterziehen, da der Nutzen schon durch die EMA-Zulassung als nachgewiesen gilt. Dies erlaubt somit in den meisten Fällen eine freie Preisgestaltung bis zu einer regulären Bewertung, die bis zu neun Jahre nach Marktzugang dauern kann [3].
Limitationen der Zulassung als Orphan Drug
Die EU-Regulierung verwendet nur die Prävalenz als Kriterium für die Anerkennung einer seltenen Krankheit. Dies hat den Nachteil, dass auch Subklassen von z. B. hämatologischen Krankheiten als „selten“ eingeordnet werden können. Die European Organisation for Rare Diseases (EURORDIS) hat in einer Stellungnahme daher angeregt, auch eine Inzidenz von 5/10.000 zu berücksichtigen – und damit auch die Anzahl der neuen Diagnosen [4]. Das würde eine Einordnung solcher onkologischen Fälle einer Second-Line-Therapie erschweren, da die Inzidenz der Diagnose und nicht nur die Prävalenz der Subgruppen in der Bewertung berücksichtigt würde. Die Förderung nach der Direktive 141/2000 würde somit eher den Patienten zugutekommen, die z. B. sehr seltene genetische Defekte haben, die sich meistens in der Kindheit manifestieren. In der gleichen Stellungnahme regt EURORDIS auch eine differenziertere Förderung an, da nach der jetzigen Regulierung nicht ausreichend unterschieden wird, ob eine Krankheit selten, wie Subklassen des B-Zell-Lymphoms mit Patientenzahlen in Europa von mehr als 200.000 [5], oder sehr selten wie z. B. das Barraquer-Simons-Syndrom mit etwa 100 Fällen, ist [5].
Vorteil muss nachweisbar sein
Die Direktive 141/2000 ist ohne Zweifel ein wichtiger Schritt zur Verbesserung der problematischen Situation von weltweit Millionen von Patientinnen und Patienten, die unter Krankheiten leiden, die wegen der Seltenheit für die pharmazeutische Industrie wirtschaftlich unattraktiv sind. Allerdings zeigt sich auch, dass diese Maßnahme noch nicht den gewünschten Erfolg erreicht hat. Nach Angaben der WHO gibt es zwischen 5000 und 8000 bekannte seltene Krankheiten, die meistens auf genetischen Defekten basieren und in Europa ca. 30 Millionen Patientinnen und Patienten betreffen [6]. Nach Angaben der European Organisation for Rare Diseases besteht allerdings nur für etwa 6% davon eine therapeutische Option [7]. Auch wenn in vielen Fällen die Komplexität der Krankheit eine Behandlung erschwert, so bleibt die Situation trotzdem unbefriedigend. Darüber hinaus ist es auffällig, dass hämatologische und onkologische Indikationen mehr als ein Drittel der zugelassenen Orphan Drugs in Europa ausmachen. Es handelt sich in vielen Fällen um Patientinnen und Patienten, die bereits eine oder sogar mehrere Therapien in der Indikation erfolglos abgeschlossen haben. Die Vermutung, dass die auf der Prävalenz basierende Definition einer seltenen Krankheit eine künstliche Klassifizierung von lukrativen hämatoonkologischen Erkrankungen erlaubt, ist daher keinesfalls unbegründet [4].
Auch wenn es wünschenswert ist, Therapien für seltene Krankheiten überhaupt zur Verfügung zu haben, so ist es aber auch – besonders in Anbetracht der meistens sehr hohen Kosten – legitim, die Frage zu stellen, wie effektiv diese sind, das heißt welche Vorteile sie in der realen Welt gegenüber vorhandenen therapeutischen Alternativen haben. An dieser Stelle sei daran erinnert, dass die EMA eine Zulassung nicht verweigern kann, wenn die eingereichten Daten die Wirksamkeit und Sicherheit des Arzneimittels beweisen. Eine Zulassung erfolgt also unabhängig davon, ob andere Therapien für die Indikation vorhanden sind. Ein zugelassener Wirkstoff muss daher nicht unbedingt besser sein als bereits etablierte Alternativen.
Die Ergebnisse einer Untersuchung von 15 zwischen 2000 und 2017 anerkannten und als Fertigarzneimittel in Europa zugelassenen Orphan Drugs im onkologischen Bereich sind diesbezüglich ernüchternd [8]: Obwohl die Zulassungsstudien meistens Verbesserungen in den Surrogat-Parametern und somit auch eine Wirksamkeit der Arzneistoffe gezeigt hatten, war bei 40% der Therapien die Überlebenszeit (Overall Survival, OS) kürzer als drei Monate, was eine sehr geringe Effektivität in der realen Welt bedeutet.
In diesem Zusammenhang ist auch die vor Kurzem vom IQWiG publizierte kritische Stellungnahme interessant [3]: In dieser bemängelt das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen, dass durch den Orphan-Drug-Status eine reguläre Bewertung gegenüber einer zweckmäßigen Vergleichstherapie entfällt. Wird diese später wegen der Überschreitung der Jahresumsatzgrenze von 50 Millionen Euro oder durch den Verlust des Orphan-Drug-Status jedoch durchgeführt, so ergibt sich nur für 22% der Therapien ein beträchtlicher und für 2% ein erheblicher Zusatznutzen. Diese Befunde sind enttäuschend. Die nicht unbedeutende Entlastung der Pharmaindustrie durch die Direktive 141/2000 sollte zu besseren Ergebnissen in der realen Umgebung führen, und nicht zu solchen, die dem Kriterium eines „erheblichen Nutzens“, wie in der Direktive formuliert, kaum genügen.
Auch die Frage nach den Kosten solcher Therapien ist nicht immer abzuweisen: Eine nachvollziehbare Begründung, warum Therapien, die bereits off label für wenige Euro verschrieben wurden, nach einer Zulassung als Orphan Drug das Mehrfache kosten, fehlt in vielen Fällen. Es kann nicht der Zweck der Förderung sein, Therapien zur Verfügung zu stellen, die vom Gesundheitswesen wegen der inadäquaten Kosten nicht getragen werden können.
Das International Rare Diseases Research Consortium (IRDiRC) hat in einer aktuellen Publikation einige interessante Gedanken ausgearbeitet, wie die Forschung zu seltenen Krankheiten nicht nur in Europa effizienter gestaltet werden könnte [7]: Bis 2027 sollen weitere 1000 Therapien erforscht und implementiert werden, die als Fokus diejenigen Krankheiten haben, für die momentan keine andere Option besteht. Das IRDiRC hat dafür ein Handbuch entwickelt [9], das einen Überblick der notwendigen Schritte bis zur Zulassung beschreibt und als Hilfe für akademische Einrichtungen und kleinere Firmen dienen soll.
Zusammenfassend kann man sagen, dass vieles für die Therapie von seltenen Krankheiten seitens der Politik gemacht worden ist, dass es aber auch wichtig ist, dass in Zukunft viel mehr Arzneimittel für noch nicht behandelbare Krankheiten entwickelt werden. Lücken in der Regulierung dürfen nicht dazu führen, dass Bereiche gefördert werden, die für die Pharmaindustrie attraktiv sind, aber in der Effektivität nicht die klinischen Ziele erreichen, die für die Patientinnen und Patienten relevant sind. |
Literatur
[1] Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates vom 16. Dezember 1999 über Arzneimittel für seltene Leiden. Amtsblatt Nr. L 018 vom 22/01/2000 S. 0001 – 0005, https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:32000R0141&from=DE
[2] Community Register of orphan medicinal products. Public Health – Union Register of medicinal products, http://ec.europa.eu/health/documents/community-register/html/alforphreg.htm.
[3] Evidenz zu Orphan Drugs. IQWiG-Berichte – Nr. 1269, Stand: 23. Dezember 2021, Informationen des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG), www.iqwig.de/download/ga21-01_evidenz-zu-orphan-drugs_arbeitspapier_v1-0.pdf
[4] Arzneimittel für Kinder und für seltene Krankheiten – aktualisierte Vorschriften. https://ec.europa.eu/info/law/better-regulation/have-your-say/initiatives/12767-Arzneimittel-fur-Kinder-und-fur-seltene-Krankheiten-aktualisierte-Vorschriften_de
[5] Zugelassene Orphan Drugs. Informationen des Verbands Forschender Arzneimittelhersteller e. V. (vfa), Stand: 16. Februar 2022, www.vfa.de/de/arzneimittel-forschung/datenbanken-zu-arzneimitteln/orphan-drugs-list
[6] Rare diseases. Priority diseases and reasons for inclusion, www.who.int/medicines/areas/priority_medicines/Ch6_19Rare.pdf
[7] Hivert V, Jonker AH, O’Connor D, Ardigo D. IRDiRC: 1000 new rare diseases treatments by 2027, identifying and bringing forward strategic actions. Rare Dis Orphan Drugs J 2022;1:3, http://dx.doi.org/10.20517/rdodj.2021.02
[8] Schuller Y, Biegstraaten M, Hollak CEM et al. Oncologic orphan drugs approved in the EU – do clinical trial data correspond with real-world effectiveness? Orphanet J Rare Dis 2018;13:214, https://doi.org/10.1186/s13023-018-0900-
[9] Orphan Drug Development Guidebook (ODDG). Informationen des International Rare Diseases Research Consortium (IRDiRC), https://orphandrugguide.org/



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.