














- DAZ.online
- DAZ / AZ
- DAZ 45/2021
- Korrekturen im Buch des ...

Foto: Andrey Popov/AdobeStock
Epigenetik
Korrekturen im Buch des Lebens
Das Verständnis epigenetischer Prozesse beflügelt auch die Arzneimitteltherapie
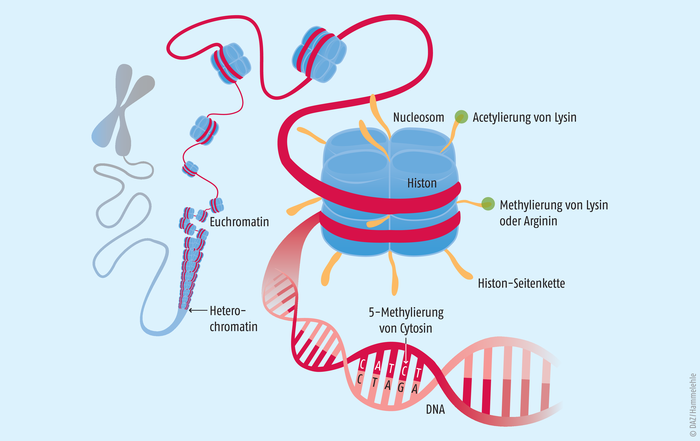
Die DNA dient den Zellen als riesiger Informationsspeicher. Sie kann somit als „Buch des Lebens“ angesehen werden. Damit sich jedoch im Verlauf der Entwicklung und Differenzierung verschiedene Organe ausbilden können, müssen bestimmte Gene aus dem gesamten Genom zellspezifisch aktiviert oder inaktiviert werden. Diese Gene bestimmen dann den Phänotyp. Die Epigenetik bestimmt, wie diese Vorgänge reguliert und an die Tochterzellgeneration weitergegeben werden. Epigenetische Vorgänge umfassen (siehe Abb. 1):
- nicht proteinkodierende RNA
- die Methylierung der DNA-Base Cytosin
- Modifikation von Histon-Proteinen im Chromatin (enthält DNA und Proteine in einer stark aufgewickelten Struktur)
Abb. 1: Ein Chromosom mit immer stärker vergrößerten Abschnitten des Chromatinfadens (DNA). Histone sind Strukturproteine, in denen die DNA teilweise nur mäßig verknäuelt ist, sodass einzelne Gene abgelesen und exprimiert werden können.Je acht Histone bilden ein Nucleosom. Enzyme, die die Histone oder die Base Cytosin in der DNA modifizieren, regulieren epigenetische Prozesse.
Dabei werden Enzyme, die eine epigenetische Modifikation einführen, auch als Writer (Schreiber) bezeichnet (z. B. Methyltransferasen).
Bei Enzymen, die Modifikationen abspalten, spricht man von Erasern (Radierer, z.B. Demethylasen) und bei Proteinen, die Modifikationen erkennen, von Readern (Leser).
Epigenetische Modifikationen beeinflussen die Transkription bestimmter Gene. Damit ein Zellverbund seine Eigenschaften beibehalten kann, müssen Modifikationen über die Zellteilung hinweg stabil sein. Im Gegensatz zur DNA ist die epigenetische Information allerdings alles andere als starr. Sie kann durch verschiedene äußere Faktoren wie Ernährung, Infektionen oder soziale Interaktionen beeinflusst werden [1]. Bei Zellen, die sich nicht mehr mitotisch teilen (z. B. Neuronen), sind die epigenetischen Modifikationen Teil eines zellulären „Langzeitgedächtnisses“. Das bedeutet: bestimmte vorübergehende Noxen können Jahre später zu zellulären Schäden führen.
Bei verschiedenen Erkrankungen konnten Abweichungen vom epigenetischen „Normalzustand“ nachgewiesen werden. Hierzu gehören unter anderem kardiovaskuläre, metabolische und neurologische Erkrankungen, aber auch Krebs [2]. Im Gegensatz zur permanenten genetischen Mutation sind sogenannte Epimutationen per se reversibel und können mittels Arzneistoffen adressiert werden. Beispielsweise kann die unterdrückte Expression eines Tumorsupressorgens wieder aktiviert werden und die Progression des Tumors vermindern. Mehrere epigenetische Mechanismen konnten auf molekularer Ebene aufgeklärt werden. Hier werden im Folgenden diejenigen vorgestellt, die bereits Ziel pharmakologischer Interventionen sind.
Methylierung bringt DNA zum Schweigen
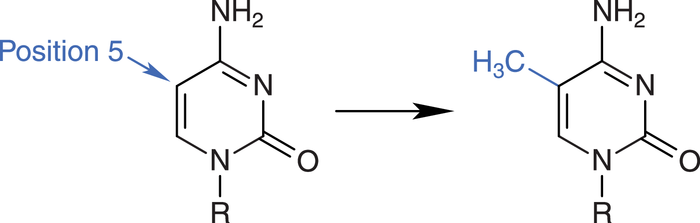
Die DNA kann durch DNA-Methyltransferasen (DNMT) methyliert werden. Dabei wird die DNA-Base Cytosin in Cytosin-Guanin-Dinucleotiden (CpG) modifiziert (Abb. 2). Sogenannte CpG-Inseln, mit einem relativ hohen Anteil des Cytosin-Guanin-Motivs, finden sich vor allem in Promotorregionen von bestimmten Genen [3].
Abb. 2: In der DNA methylieren DNA-Methyltransferasen Cytosin-Moleküle an Position 5.
Die Methylierung der DNA beeinflusst die Basenpaarung von Cytosin und Guanin im Doppelstrang nicht, sodass der genetische Code erhalten bleibt. Allerdings kommt es zu Wechselwirkungen mit Lese-Proteinen (Reader). Die Reader erkennen die epigenetische Modifikation und beeinflussen die Transkription, zusätzlich wirken sie auf die Chromatinstruktur ein.
Die DNA-Methylierung ist typischerweise mit der Stilllegung eines Gens assoziiert. Dies ist essenziell bei der Zelldifferenzierung, kann aber bei Fehlregulation zur Entstehung von Krankheiten beitragen [4]. Eine fehlgesteuerte Hypermethylierung in der Promotorregion von Tumorsuppressorgenen kann zur Tumorgenese beitragen. Daher stellen DNA-Methyltransferasen einen Angriffspunkt in der Krebstherapie dar.
Wirkstoffe hemmen DNA-Methyltransferasen und aktivieren Gene
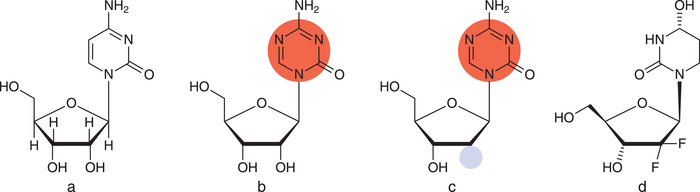
Mit Azacitidin (Vidaza®) ließ die US-amerikanische Arzneimittelbehörde FDA 2004 erstmalig einen Arzneistoff mit epigenetischem Target zu. Zwei Jahre später folgte die Zulassung für Decitabin (Dacogen®), ein Desoxyribose-Analogon von Azacitidin (s. Abb. 3). Beide Stoffe stellen Cytidin-Derivate dar, jedoch wurde im Pyrimidin-Ring ein zusätzliches Stickstoffatom eingebaut. Zunächst wird der Wirkstoff metabolisch aktiviert und in die DNA eingebaut, Azacitin zudem in die RNA. Anschließend erkennt die DNA-Methyltransferase die Wirkstoffe aufgrund ihrer Ähnlichkeit zu Cytidin, dem natürlichen Substrat. Jedoch hemmt das Substrat das Enzym kovalent und irreversibel [5]. In der Folge verschiebt sich das Gleichgewicht: Die vormals hypermethylierten – und damit „stillgelegten“ – Gene werden nun vermehrt in Proteine übersetzt. Der Effekt ist allerdings unselektiv und findet damit im gesamten Genom statt [6].
Abb. 3: a: Cytidin (RNA-Baustein); b: Azacitidin; c: Decitabin; markiert ist der modifizierte Pyrimidin-Ring (rot) sowie die Desoxyribose (blau); d: Cytidin-Desaminase Hemmer Cedazuridin
Sowohl Azacitidin als auch Decitabin besitzen darüber hinaus zytotoxische Effekte, die auch zum Therapieeffekt beitragen. Ihre Entwicklung geht bis in die 1950er-Jahre zurück. Damals rückten Nukleosid-Derivate als vielversprechende Zytostatika ins Zentrum der Forschung. Erst später erkannte man, dass die Nukleoside die DNA-Methyltransferase hemmen können – und das bereits bei geringen Plasmakonzentrationen.
Azacitidin ist Mittel der Wahl bei der Behandlung des myelodysplastischen Syndroms (MDS), sofern keine Stammzelltransplantation möglich ist. Es kann auch bei der chronisch myelomonozytären Leukämie (CMML) und bei akuter myeloischer Leukämie (AML) angewendet werden. Decitabin ist bei AML indiziert. Sowohl Azacitidin als auch Decitabin weisen ein ungünstiges pharmakokinetisches Profil auf. Ihre Bioverfügbarkeit ist schlecht, sie sind physikochemisch instabil und werden zudem durch die Cytidin-Desaminase abgebaut. Um die Bioverfügbarkeit der DNA-Methyltransferase-Inhibitoren im Körper zu steigern, wurde mit Cedazuridin ein Hemmstoff der Cytidin-Desaminase entwickelt. Die FDA ließ 2020 eine Fixkombination mit Decitabin zu (Inqovi®) [7].
In den Fokus klinischer Studien rücken Kombinationen epigenetischer Arzneistoffe mit anderen Krebstherapeutika. Vielversprechend erscheint z. B. die gemeinsame Gabe von Azacitidin mit dem Bcl-2-Hemmer Venetoclax (Venclyxto®) [8]. Bcl-2 (B-cell-lymphoma-2) ist ein Protein mit anti-apoptotischer Wirkung, das in Krebszellen den programmierten Zelltod verhindert – und so zur Hyperproliferation dieser Zellen beiträgt. Venetoclax inhibiert Bcl-2 und setzt dabei gleichzeitig zuvor gebundene pro-apoptotische Proteine frei, die ihrerseits den kontrollierten Zelltod stimulieren.
2021 ließ die Europäische Arzneimittelagentur (EMA) die Kombination Azacitidin bzw. Decitabin/Venetoclax zu – für AML-Patienten, bei denen eine Therapie mit intensiver Chemotherapie nicht möglich ist.
Locker oder dicht gepackt? Ein Gleichgewicht
Die DNA einer menschlichen Zelle besitzt eine Gesamtlänge von etwa 2 m. Sie muss eng im nur 10 bis 100 µm großen Zellkern verpackt werden. Hierzu wird die DNA um basische Histon-Octamer-Proteine gewickelt. Der gebildete Komplex aus DNA und Histonen wird als Nucleosom bezeichnet. Durch die Aneinanderreihung mehrerer dieser Nucleosome bildet sich eine Chromatin-Faser, welche vollständig kondensiert die Chromosomen bilden. Das Chromatin kann in zwei Formen vorliegen:
- Euchromatin, welches vergleichsweise locker gepackt vorliegt und leichter zugänglich ist für z. B. Transkriptionsfaktoren oder die RNA-Polymerase und
- Heterochromatin, welches dichter gepackt und transkriptionell eher inaktiv ist [3].
Die beiden Strukturen können sich dynamisch ineinander umwandeln – ein Prozess, der die Genregulation stark beeinflusst. Ihn steuert die post-translationale Modifikation der Histone. Sie werden unter anderem acetyliert und methyliert (s. u.) und durch Kinasen phosphoryliert. Auch Ubiquitinylierungen oder ADP-Ribosylierungen wurden beschrieben [9].
Enzyme (des)acetylieren Histone – ein Wirkstofftarget
Acetylreste aus dem Coenzym Acetyl-CoA können kovalent freie Aminogruppen der Lysin-Seitenketten der Histone binden – katalysiert durch die Histon-Acetyltransferasen (HAT). Acetylierte Histone interagieren weniger stark mit DNA. Die Chromatin-Struktur lockert sich auf und wird zum Euchromatin, die Transkription wird aktiv. Die Acetylgruppen können wiederum von den Histonen abgespalten werden. Histon-Desacetylasen (HDAC) katalysieren diesen Prozess. Sind Histon-Desacetylasen übermäßig aktiv, ist das unter anderem mit einer verminderten Expression von Tumorsuppressor-Genen assoziiert. Daher bot es sich an, Hemmstoffe dieses Enzyms zu entwickeln.
Histon-Desacetylasen werden in vier Klassen eingeteilt, die zwei unterschiedliche katalytische Mechanismen aufweisen. Die HDAC der Klassen I, II und IV hydrolysieren das Amid in der Seitenkette der Aminosäure Lysin Zink-abhängig. Dagegen spalten Histon-Desacetylasen der Klasse III die Acetylgruppe abhängig vom Coenzym NAD+.
Histon-Acetyltransferasen und -Desacetylasen modifizieren in der Zelle nicht nur Histone, sondern auch andere Proteine, z. B. Tubulin in den Mikrotubuli oder den Tumorsuppressor p53 [1]. Noch ist unklar, welche Rolle dies für den klinischen Erfolg der Hemmstoffe spielt.
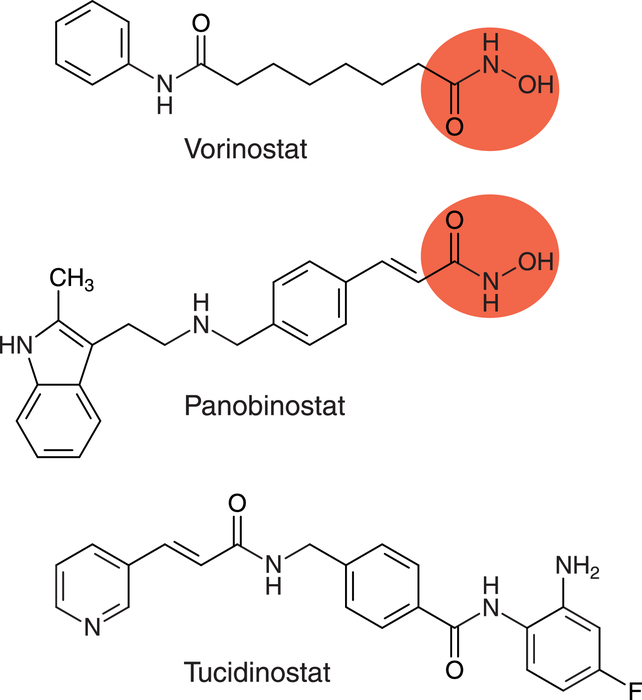
Abb. 4: Strukturen einiger wichtiger HDAC-Inhibitoren, rot markiert ist die Hydroxamsäurefunktion
Erstmals ließ die FDA 2006 mit Vorinostat (Zolinza®) einen Histon-Desacetylase-Inhibitor (HDAC) zu (s. Abb. 4.) Im Wirkstoff-Molekül komplexiert die Hydroxamsäure am Ende der Alkylkette den Cofaktor Zink der HDAC-Klassen I, II und IV (pan-HDAC-Inhibitor). Das selbe Strukturmotiv findet sich auch in den Derivaten der Zimtsäure Panobinostat (Farydak®) und Belinostat (Beleodaq®), welche ebenfalls unselektiv Zink-abhängige Histon-Desacetylasen hemmen. Typische Nebenwirkungen bei der Therapie sind Durchfall, Erbrechen und Thrombozytopenie. Die drei Arzneistoffe sind durch die FDA und EMA für folgende Indikationen zugelassen:
- Vorinostat: kutanes T-Zell-Lymphom (CTCL), Mittel der dritten Wahl
- Panobinostat: multiples Myelom (in Kombination mit dem Proteasominhibitor Bortezomib und Dexamethason)
- Belinostat: peripheres T-Zell-Lymphom (PTCL)
Selektive HDAC-Hemmer in der Pipeline
Romidepsin (Istodax®) ist ein selektiver Inhibitor der HDAC-Klasse I. Das cyclische Peptid wurde aus dem Bakterium Chromobacterium violaceum isoliert. Nach metabolischer Aktivierung interagiert es über eine freigesetzte Thiolfunktion mit dem Zink-Ion der Histon-Desacetylase und hemmt so das Enzym [10]. Wie Vorinostat wird Romidepsin beim kutanen T-Zell-Lymphom angewendet. Es erhielt jedoch keine Zulassung durch die EMA, da diese den Zusatznutzen als nicht ausreichend belegt ansah [11].
Tucidinostat (Epidaza®) wurde 2015 in China für die Therapie des refraktären und rückfälligen peripheren T-Zell-Lymphoms zugelassen. Das Benzamid hemmt vor allem Klasse I der Histon-Desacetylasen [12]. Tucidinostat ist der erste moderne Arzneistoff, der vollständig in China entwickelt wurde. Eine Studie aus dem Jahr 2019 legt nahe, dass postmenopausale Patientinnen mit hormontherapieresistentem Mammakarzinom von der Kombination aus Tucidinostat und einem Aromatasehemmer (Exemestan) profitieren könnten. Hierzu stehen allerdings noch weitere Untersuchungen aus [13].
Zahlreiche weitere HDAC-Inhibitoren befinden sich in der Pipeline der Pharmaindustrie. Das Hauptaugenmerk liegt darauf, selektive Hemmstoffe für die einzelnen HDAC-Subtypen zu entwickeln, um eine gezieltere Therapie zu ermöglichen. Darüber hinaus rücken auch Kombinationstherapien mit lang erprobten Zytostatika in den Fokus, auch um gegebenenfalls die Dosis der teils stark toxischen Substanzen zu reduzieren und Nebenwirkungen zu mindern.
Dass der Einsatz nicht auf die Krebstherapie begrenzt sein muss, zeigt Givinostat, ein pan-HDAC-Inhibitor. Aktuell befindet Givinostat sich in einer Phase-III-Studie zur Behandlung der Muskeldystrophie des Typs Duchenne (DMD).
Den Methylgruppen-Transfer bremsen
Histone können an den basischen Aminosäuren Lysin und Arginin methyliert werden. Lysin wird dabei mit bis zu drei Methylgruppen versehen. Den Methylgruppen-Transfer katalysiert die Histonmethyltransferase (HMT). Die Methylierung kann Gene aktivieren aber auch hemmen – abhängig von der Position des Lysins im Histon und vom zellulären Kontext [1].
Tazemetostat (Tazverik®) hemmt selektiv die EZH2-Histonmethyltransferase. In verschiedenen Krebsarten ist das EZH2-Gen überexprimiert, was mit einer schlechten Prognose assoziiert ist [14]. Tazemetostat ist der erste zugelassene Hemmstoff einer Histonmethyltransferase überhaupt. Anfang 2020 gab die FDA in einem beschleunigten Verfahren grünes Licht für den Wirkstoff zur Behandlung von metastasierendem bzw. lokal fortgeschrittenem epithelialen Sarkom für Personen über 16 Jahren [15]. Damit ist der Wirkstoff das erste epigenetische Therapeutikum, das bei einem soliden Tumor indiziert ist.
Im Juni 2020 wurde auch eine Zulassung zur Therapie von refraktärem und rezidivem follikulärem Lymphom bei Erwachsenen mit EZH2-Mutation durch die FDA erteilt [16]. In den Zulassungsstudien traten vornehmlich Nebenwirkungen wie Übelkeit, Obstipation und verminderter Appetit auf. Doch es gab auch Hinweise auf ein gesteigertes Risiko von Sekundärtumoren sowie einer potenziellen Teratogenität [15]. Aktuell laufen weitere Studien zur Anwendung von Tazemetostat. Dabei testen die Forscher auch mögliche Kombinationen mit bereits etablierten Substanzen [17].
Histon-Demethylaseinhibitoren ähneln MAO-Hemmern
Auch die Lysin-spezifische Demethylase 1 (LSD1) modifiziert Histone epigenetisch [14]. Sie ähnelt strukturell den MAO-Enzymen, die als Arzneistoff-Targets in der Therapie von Depressionen oder Morbus Parkinson adressiert werden. Der unselektive und irreversible Inhibitor der Monoaminoxidase (MAO) Tranylcypromin blockiert auch LSD1 in klinisch erreichbaren Konzentrationen [1]. Klinische Studien, in denen man Tranylcypromin mit anderen Krebstherapeutika kombinierte, lieferten jedoch eher mäßige Erfolge. Auf Grundlage von Tranylcypromin wurden potente und selektive Wirkstoffe entwickelt, die Lysin-spezifische Demethylase 1 irreversibel hemmen. Diese befinden sich aktuell in der klinischen Prüfung. Einige Substanzen könnten in naher Zukunft den Sprung zur Marktzulassung schaffen und das Anwendungsfeld epigenetischer Wirkstoffe über Krebs hinaus erweitern. Einige ausgewählte Beispiele [18]:
- Iadademstat: Phase II bei akuter myeloischer Leukämie und kleinzelligem Lungenkarzinom (SCLC)
- Bomedemstat: Phase II zur Therapie von essenzieller Thrombozythämie und Myelofibrose
- Vafidemstat: Phase II zur Therapie von Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS), Borderline-Persönlichkeitsstörung, Schizophrenie (negativ Symptomatik) und der Alzheimer-Krankheit
Vafidemstat ist ein Inhibitor der Lysin-spezifischen Demethylase 1, der die Blut-Hirn-Schranke gut überwindet. Weil er auch MAO-B-Isoenzyme hemmt, rückte seine Anwendung im psychiatrischen und neurologischen Bereich in den Fokus. Auch zeigt der Wirkstoff einen antiinflammatorischen Effekt. Hierdurch ergeben sich Anwendungsmöglichkeiten bei Erkrankungen mit übermäßigen Immunreaktionen, wie multiple Sklerose oder COVID-19. Die vorläufigen Ergebnisse einer Phase-II-Studie zeigten kürzlich, dass die Überreaktion des Immunsystems bei Patienten mit schwerem COVID-19-Verlauf durch Vafidemstat gedrosselt werden kann [19].
Diese drei Wirkstoffe basieren strukturell auf Tranylcypromin und hemmen die Lysin-spezifische Demethylase 1 irreversibel. Daneben wurden inzwischen auch reversible Hemmstoffe entwickelt wie Seclidemstat oder CC-90011. Auch sie werden derzeit klinisch geprüft [20, 21].
Wenn Metabolite auf epigenetische Radierer einwirken
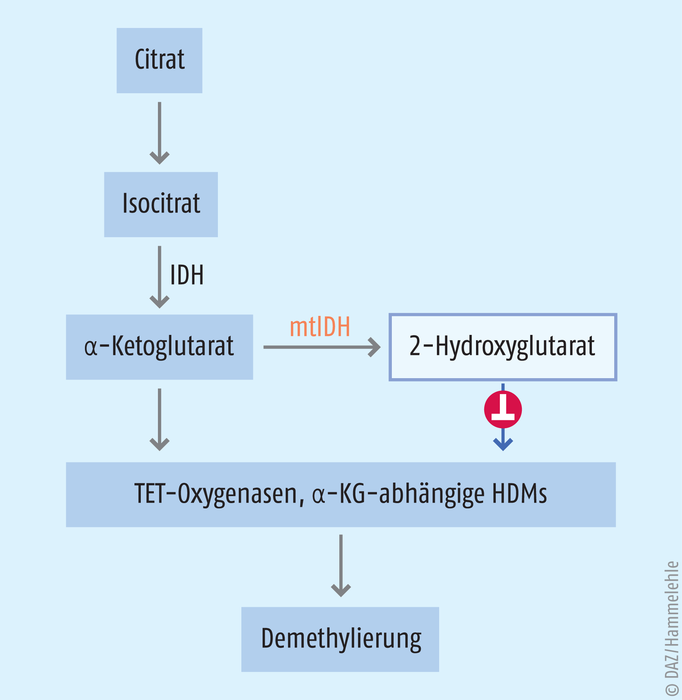
Enzyme, die Fe(II)-abhängig mit dem Cofaktor α-Ketoglutarat Methylgruppen oxidieren, führen zur Demethylierung von Histonen und der DNA. Histon-Demethylasen (HDM) spalten Formaldehyd ab, wohingegen die sogenannten TET-Oxygenasen Methylcytosin in der DNA stufenweise zu Hydroxymethyl-, Formyl- und Carboxyl-Cytosin umwandeln (s. Abb. 5). Anschließend erfolgt ein Austausch gegen Cytosin mittels Basenexzisionsreparatur [3].
Abb. 5: α-Ketoglutarat (α-KG) entsteht nach Isocitrat-Dehydrogenase(IDH)-Katalyse aus Isocitrat. Die mutierte IDH (mtIDH) kann den Metaboliten 2-Hydroxyglutarat bilden. 2-Hydroxyglutarat wiederum hemmt α-KG-abhängige Histondemethylasen (HDM) und TET-Oxygenasen. In der Folge bleibt die DNA stärker methyliert und bestimmte Gene bleiben stumm.
Im menschlichen Organismus synthetisiert die Isocitrat-Dehydrogenase (IDH) α-Ketoglutarat aus Isocitrat. Bei akuter myeloischer Leukämie kann bei bis zu 30% der Patienten eine Gain-of-function-Mutation der IDH nachgewiesen werden. Bei einer Gain-of-function-Mutation gewinnt das betreffende Enzym eine neue Eigenschaft hinzu. In diesem Fall hat die Mutation pathophysiologische Folgen: Die mutierte Isocitrat-Dehydrogenase reduziert α-Ketoglutarat weiter zum 2-Hydroxyglutarat [22]. Dieser Metabolit hemmt die Enzyme, die von α-Ketoglutarat abhängen. Damit stabilisiert der Metabolit die Hypermethylierung an Histone oder die DNA – mit den oben beschriebenen Folgen: bestimmte Gene werden stillgelegt, schlimmstenfalls jene, die unkontrollierte Zellteilungen unterdrücken. Wissenschaftler entwickelten Moleküle, die mutierte Isocitrat-Dehydrogenasen (mtIDH) selektiv hemmen: Enasidenib (Idhifa®) und Ivosidenib (Tibsovo®). Die FDA ließ die Substanzen 2017 bzw. 2018 bei refraktärer und wiederkehrender akuten myeloischen Leukämie zu. Für Ivosidenib wurde 2019 die Zulassung nochmals erweitert. Der Wirkstoff ist seitdem Mittel der ersten Wahl bei AML-Patienten, bei denen eine Standardtherapie nicht in Frage kommt. In den Zulassungsstudien zeigte sich, dass sich die Lebensqualität der Behandelten verbessert hatte. Die Indikation gilt aber nur, wenn vorher nachgewiesen wird, dass die Isocitrat-Dehydrogenase mutiert ist.
Die durch die Inhibitoren der Isocitrat-Dehydrogenase (IDH-Hemmer) induzierte Demethylierung führt zu einer Zelldifferenzierung. Bei etwa 20% der Patientinnen und Patienten differenzieren sich die Leukämie-Zellen während der Therapie. Das kann zu einem sogenannten Differenzierungssyndrom führen, charakterisiert durch eine starke Entzündungsreaktion, die mit Glucocorticoiden eingedämmt werden kann [22]. Auch im Falle der beiden IDH-Hemmer Enasidenib und Ivosidenib richtet sich der Fokus in der klinischen Forschung auf die Suche nach idealen Kombinationspartnern in der Therapie. Unter anderem wird auch eine Kombination mit den oben erwähnten Venetoclax und Azacitidin untersucht.
Wie es weitergehen könnte
Mitte der 1990er-Jahre entschlüsselte man zentrale epigenetische Vorgänge auf molekularer Ebene. 25 Jahre später haben einige Prinzipien zu erfolgreichen Arzneimitteln geführt und viele weitere befinden sich in klinischer Prüfung. Auch wenn die Anwendungen bisher in aller Regel auf wenige Krankheiten mit relativ wenigen Patientinnen und Patienten beschränkt sind – es gibt Hoffnung, dass in Zukunft breitere Indikationen folgen. Vor allem werden selektivere Hemmstoffe entwickelt und nach Kombinationen epigenetischer Arzneimittel mit etablierteren Substanzen gesucht. Das Feld der Epigenetik ist dynamisch und wird weiterhin spannende Erkenntnisse liefern, die (hoffentlich) therapeutische Optionen eröffnen, die über die Krebstherapie hinausgehen können. |
Literatur
[1] Wenzler S, Jung M. Lesezeichen im Buch des Lebens – Gezielte Modifikationen machen die Genexpression der Krebszellen angreifbar. Deutsche Apotheker Zeitung 2017;44:60
[2] Heerboth S et al. Use of epigenetic drugs in disease: an overview. Genet Epigenet 2014, doi: 10.4137/GEG.S12270
[3] Knippers R et al. Molekulare Genetik. 11. Auflage, Thieme Verlag, Stuttgart 2018:443
[4] Meissner A et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008;454(7205):766-770
[5] Ganesan A et al. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenetics 2019, doi.org/10.1186/s13148-019-0776-0
[6] Berdasco M et al. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet 2019, doi:10.1038/s41576-018-0074-2
[7] Patel A et al. Cedazuridine/decitabine: from preclinical to clinical development in myeloid malignancies. Blood Adv 2021, doi.org/10.1182/bloodadvances.2020002929
[8] Lübbert M et al. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes. Leukemia 2021, doi.org/10.1038/s41375-021-01218-0
[9] Kouzarides T et al. Chromatin modifications and their function. Cell 2007, doi:10.1016/j.cell.2007.02.005
[10] Van der Molen K et al. Romidepsin: a natural product recently approved for cutaneous T-cell lymphoma. Antibiot 2011, doi.org/10.1038/ja.2011.35
[11] Istodax: Übersicht der European Medicines Agency (EMA), www.ema.europa.eu. Stand: 23. August 2021
[12] Pan D et al. Discovery of an orally active subtype-selective HDAC inhibitor, chidamide, as an epigenetic modulator for cancer treatment. Med Chem Commun 2014, doi.org/10.1039/C4MD00350K
[13] Romero D. HDAC inhibitors tested in phase III trial. Nat Rev Clin Oncol 2019, doi:10.1038/s41571-019-0224-2
[14] Cheng Y et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Nature 2019, doi.org/10.1038/s41392-019-0095-0
[15] Hoy S. Tazemetostat: First Approval. Nature Drugs 2020, doi:10.1007/s40265-020-01288-x
[16] FDA granted accelerated approval to tazemetostat for follicular lymphoma. Mitteilung der Food and Drug Administration (FDA), www.fda.gov, Stand: 7. August 2021
[17] Italiano A. Targeting epigenetics in sarcomas through EZH2 inhibition. J Hematol Oncol 2020, doi:10.1186/s13045-020-00868-4
[18] Fang Y et al. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol 2019, doi.org/10.1186/s13045-019-0811-9
[19] Oryzon presents safety and efficacy data of vafidemstat from the Phase II ESCAPE trial in severe COVID-19 patients at ECCMID-2021. Pressemitteilung von Oryzon Genomics, Stand: 12. August 2021
[20] Kanouni T et al. Discovery of CC-90011: A Potent and Selective Reversible Inhibitor of Lysine Specific Demethylase 1 (LSD1). J Med Chem 2020, doi:10.1021/acs.jmedchem.0c00978
[21] Reed DR et al. Phase 1 trial of seclidemstat (SP-2577) in patients with relapsed/refractory Ewing sarcoma. J Clin Oncol 2021, doi:10.1200/JCO.2021.39.15_suppl.11514
[22] McMurry H et al. D-2-Hydroxyglutarate in Glioma Biology. Curr Hematol Malig Rep 2021, doi:10.3390/cells10092345
Autoren
Nicolas Heller studiert seit 2017 Pharmazie in Freiburg und wird im Jahr 2022 bei Prof. Dr. Manfred Jung das Pharmazeutische Praktikum im Bereich Entwicklung epigenetischer Hemmstoffe im Rahmen einer Diplomarbeit absolvieren.
Prof. Dr. Manfred Jung studierte Pharmazie an der Universität Marburg und promovierte 1993 bei Prof. Dr. Wolfgang Hanefeld. Nach einem Postdoktorat an der Universität Ottawa (Kanada) habilitierte er im Fach Pharmazeutische Chemie an der Universität Münster. Seit 2003 ist er Professor für Pharmazeutische Chemie am Institut für Pharmazeutische Wissenschaften der Universität Freiburg. Prof. Jung ist seit 2012 Co-Sprecher des Sonderforschungsbereichs SFB992 „Medical Epigenetics“ und seit 2019 Sprecher der Hochschullehrenden im Senat der Universität Freiburg. Für seine Forschung im Bereich der Entwicklung epigenetischer Wirkstoffe erhielt er den Phoenix Pharmazie Wissenschaftspreis 2016 im Fach Pharmazeutische Chemie.
Homepage: www.jungm.de, Twitter: @ChemEpi



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.