














- DAZ.online
- DAZ / AZ
- DAZ 4/2021
- Rebellion gegen ...

Foto: Jag_cz – stock.adobe.com
Nebenwirkungen
Rebellion gegen Arzneistoffe
Das Stevens-Johnson-Syndrom und die toxische epidermale Nekrolyse im Fokus
Albert Mason Stevens und Frank Chambliss Johnson beschrieben im Jahre 1922 zwei Jungen mit einem generalisierten Ausschlag, der von Fieber sowie einer schweren Entzündung der Mundschleimhaut und einer schweren Konjunktivitis begleitet war [1]. Die beiden Ärzte konnten keine eindeutige Diagnose stellen und vermuteten unter anderem eine Erkrankung bis dahin unbekannten Ursprungs. Heute trägt dieses Krankheitsbild den Namen dieser Pioniere. Das Stevens-Johnson-Syndrom (SJS) sowie die von Alan Lyell im Jahre 1956 definierte toxische epidermale Nekrolyse (TEN, medikamentös induziertes Lyell-Syndrom) sind schwere Überempfindlichkeitsreaktionen des Körpers auf Arzneimittel oder seltener auf Infektionen [2]. Vereinfacht bezeichnen beide Begriffe das gleiche Krankheitsbild (hier zusammengefasst als SJS/TEN bezeichnet), unterscheiden sich aber im Schweregrad der Erkrankung. Während beim Stevens-Johnson-Syndrom weniger als 10% der Haut betroffen sind, erstreckt sich die toxische epidermale Nekrolyse über mehr als 30% der Hautoberfläche. Im Bereich dazwischen spricht man von einer Übergangsform. Mit einer Inzidenz von ein bis zwei Fällen auf eine Million Einwohner tritt diese Reaktion zwar sehr selten, allerdings unvorhersehbar auf. Die Mortalität der Erkrankung ist hoch [2]. 1 bis 5% der Patienten mit Stevens-Johnson-Syndrom versterben, während 25 bis 35% eine toxische epidermale Nekrolyse nicht überleben. In der älteren Bevölkerung ist die Sterblichkeit noch höher [2].
Risiko Arzneimittel
Viele verschiedene Arzneistoffe wurden mit dem Auftreten des Stevens-Johnson-Syndroms und der toxischen epidermalen Nekrolyse in Verbindung gebracht (s. Kasten „Beispiele für Wirkstoffe mit bekanntem Risiko für SJS/TEN“). Einer großen multinationalen Fall-Kontroll-Studie [3] zufolge ist das Risiko vor allem bei den Antikonvulsiva Phenytoin, Carbamazepin, Phenobarbital und Lamotrigin erhöht. Eine weitere Gefahr stellen die Sulfonamid-Verbindungen Sulfamethoxazol sowie Sulfasalazin dar. Interessanterweise fanden sich für andere Sulfonamid-verwandte Verbindungen wie Thiazid-Diuretika, Furosemid und Sulfonylharnstoffe nur wenige Hinweise auf ein erhöhtes Risiko. Zu den riskanten Medikamenten zählen des Weiteren das Urikostatikum Allopurinol, der nicht-nukleosidische Reverse-Transkriptase-Inhibitor Nevirapin sowie nichtsteroidale Antirheumatika aus der Gruppe der Oxicame. Allopurinol erhöht das Risiko zudem abhängig von der verwendeten Dosis, insbesondere mehr als 200 mg täglich sind kritisch [4]. Eine geringere, aber doch signifikante Gefahr geht außerdem von den Antibiotika der Klassen der Makrolide, Chinolone, Cephalosporine, Tetracycline und Aminopenicilline sowie von den nichtsteroidalen Antirheumatika aus der Klasse der Essigsäure-Derivate wie z. B. Diclofenac aus [3]. Im Verdacht, SJS/TEN auslösen zu können, stehen zudem auch Pantoprazol und Glucocorticoide [3], jedoch ist hier die Datenlage noch nicht ausreichend. Betablocker, ACE-Inhibitoren, Calciumantagonisten, Insulin sowie die bereits erwähnten Thiazid-Diuretika, Furosemid und Sulfonylharnstoffe sind nicht mit einem erhöhten Risiko verbunden. Auch unter einer Therapie mit Checkpoint-Inhibitoren wie Ipilimumab, Nivolumab und Pembrolizumab wurden das Stevens-Johnson-Syndrom sowie die toxische epidermale Nekrolyse beschrieben [5]. Insgesamt besteht das höchste Risiko für Stevens-Johnson-Syndrom/toxische epidermale Nekrolyse vor allem in den ersten acht Wochen nach Therapiestart mit einem Medikament [3]. Neben Arzneimitteln, die für die meisten Fälle verantwortlich sind, können auch Infektionen auslösende Faktoren sein. Gerade bei Kindern liegen diese häufiger dem Stevens-Johnson-Syndrom zugrunde [6]. Ein erhöhtes Risiko, das Syndrom zu entwickeln, haben Schwangere aufgrund des geänderten immunologischen Status während der Schwangerschaft [7]. Auch HIV-Infizierte sind generell gefährdeter [2].
Beispiele für Wirkstoffe mit bekanntem Risiko für SJS/TEN
hohes Risiko
- Antikonvulsiva
- Phenytoin
- Carbamazepin
- Phenobarbital
- Lamotrigin - Sulfonamide
- Sulfamethoxazol
- Sulfasalazin - Allopurinol
- Nevirapin
- Oxicame
erhöhtes Risiko
- Antibiotika
- Makrolide
- Chinolone
- Cephalosporine
- Tetracycline
- Aminopenicilline - nichtsteroidale Antirheumatika (NSAR)
- Essigsäurederivate, z. B. Diclofenac
kein Risiko
- Betablocker
- ACE-Inhibitoren
- Calciumkanal-Antagonisten
- Insulin
- Thiazid-Diuretika
- Furosemid
- Sulfonylharnstoffe
T-Zellen im Visier
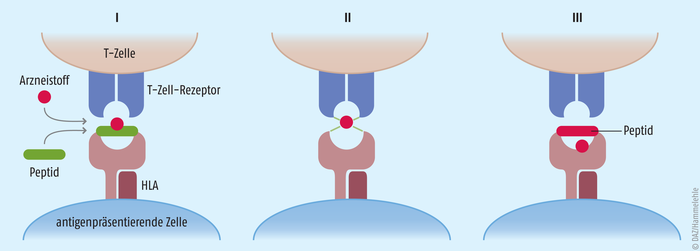
Die Pathogenese dieses Krankheitsbildes ist bislang noch nicht vollständig verstanden. Es handelt sich wahrscheinlich um eine T-Lymphozyten-vermittelte Überempfindlichkeitsreaktion, auch Typ-IV-Reaktion oder Spättypreaktion genannt. Vereinfacht ausgedrückt werden in diesem Fall T-Zellen durch Arzneistoffe aktiviert. Wie genau es dazu kommen kann, darüber gibt es verschiedene Theorien [8] (Abb. 1): (I) Der Arzneistoff oder ein Metabolit kann im Körper mit Proteinen reagieren und einen Hapten-Protein-Komplex formen. Dieser wird von antigenpräsentierenden Zellen aufgenommen, zum Hapten-Peptid-Komplex prozessiert und an deren Oberfläche mithilfe von HLA-Molekülen (humane Leukozyten-Antigene) präsentiert. Dort kann der Komplex dann von einem passenden T-Zell-Rezeptor erkannt werden. (II) Manche Arzneistoffe wie Sulfamethoxazol können die T-Zell-Rezeptoren aber auch direkt binden und diese in Gegenwart von HLA-Molekülen zusätzlich stimulieren. (III) Ein Arzneistoff kann auch in der Lage sein, spezifisch mit HLA-Molekülen zu interagieren. Der gebundene Wirkstoff beeinflusst so, welche Antigene diese Proteine binden und welche T-Zellen aktiviert werden. Es wurden verschiedene HLA-Varianten mit einem vermehrten Auftreten des SJS/TEN in Verbindung gebracht, sodass eine genetische Prädisposition möglich ist [2]. An der Pathogenese sind verschiedene T-Zell-Populationen beteiligt. Beispielsweise sezernieren cytotoxische CD8+-T-Lymphozyten Moleküle wie Fas-Ligand, Perforin oder Granulysin, die die Apoptose der Keratinozyten einleiten und damit das Absterben der Epidermis verursachen [8]. Die Bezeichnung „Spättyp-Reaktion“ kommt dabei nicht von ungefähr. Im Schnitt machen sich die Symptome erst nach vier Tagen bis vier Wochen bemerkbar, was eine eindeutige Identifizierung des auslösenden Medikaments schwierig gestalten kann.
Abb. 1: T-Zell-vermittelte zytotoxische Reaktion auf Arzneimittelantigene Arzneistoffe können einen Hapten-Peptid-Komplex bilden, der von antigenpräsentierenden Zellen (APZ) an ihrer Oberfläche präsentiert wird (I). Arzneistoffe können auch direkt T-Zell-Rezeptoren aktivieren und in Gegenwart von HLA-Molekülen (Humane Leukozyten-Antigene) zusätzlich stimulieren (II). Ein Arzneistoff kann auch spezifisch mit HLA-Molekülen interagieren. Der gebundene Wirkstoff beeinflusst so, welche Peptide diese Proteine binden und welche T-Zellen aktiviert werden (III).
Schwere Hautreaktion
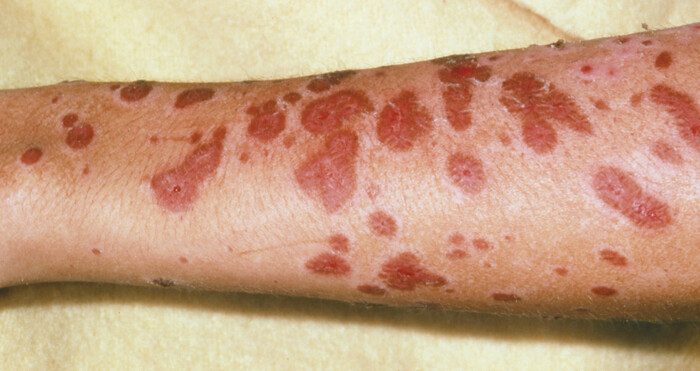
Oftmals geht der Hautreaktion ein Prodromalstadium voraus, das unter anderem von Abgeschlagenheit, Fieber sowie Kopfschmerzen und Schnupfen gekennzeichnet sein kann [9]. Auf Haut und Schleimhaut formen sich zunächst purpurne Makulae (Abb. 2) und abgegrenzte Erytheme mit zentraler dunkelroter Färbung, sogenannte atypische Kokarden. Initial betroffen sind meist der Oberkörper und das Gesicht. Die Läsionen können sich im Laufe der Erkrankung vergrößern, zusammenfließen und über den ganzen Körper ausbreiten. Oft bilden sich in betroffenen Hautarealen Blasen [9]. Bereits minimale Scherkräfte reichen aus, um die betroffene Epidermis abzulösen. Diesen Befund bezeichnet man als Nikolsky-Zeichen, welches ein diagnostisches Indiz für das Stevens-Johnson-Syndrom bzw. die toxische epidermale Nekrolyse ist. Die sich ablösende Epidermis legt die darunter liegende Dermis frei, welche nässt und zu Blutungen neigt. Die Haut ist daher anfällig für Infektionen, die häufigste Komplikation des Stevens-Johnson-Syndroms und der toxischen epidermalen Nekrolyse. Diese können in eine Sepsis münden und zum Tod führen. Durch die teils großflächige Ablösung der Epidermis ist zudem die Temperaturregulation gestört und die Barrierefunktion der Haut beeinträchtigt [9]. Der Patient verliert dadurch große Mengen an Flüssigkeit. Ein möglicher hypovolämischer Schock kann eine Nierenschädigung verursachen und muss daher verhindert werden. Häufig sind auch die Schleimhäute auf der Körperoberfläche sowie im Körperinneren beteiligt [9]. Betroffen sind vor allem die Mund- und Nasenschleimhaut sowie Augen und Genitalbereich. Aber auch die Magen-Darm-Schleimhaut und die Atemwege können geschädigt werden. Je nach Ausprägung ergeben sich so verschiedene Konsequenzen. Die Mundöffnung kann z. B. bei Beteiligung der Mundschleimhäute beeinträchtigt sein. Es können Probleme beim Harnlassen auftreten, wenn der Genitalbereich betroffen ist. Bei einer Beteiligung der Augen entwickelt sich häufig eine Bindehautentzündung bis hin zu Bindehautverklebungen und Ulzerationen der Kornea. Eine Lungenschädigung kann in der Akutphase der Erkrankung eine mechanische Beatmung erforderlich machen und auch eine Pneumonie nach sich ziehen [9].
Foto: Science Photo Library / Dr. P. Marazzi
Abb. 2: Ausschlag aus roten Blasen am Arm einer Person, die am Stevens-Johnson-Syndrom leidet. Schwere Blasenbildung der Haut und Läsionen an den Schleimhäuten der Augen, des Mundes oder der Genitalien sind Zeichen einer schweren Reaktion.
Der Schweregrad der Erkrankung wird mit dem sogenannten SCORTEN-Wert (The severity-of-illness score for TEN) abgeschätzt [10]. Dieser wird innerhalb der ersten 24 Stunden nach der stationären Aufnahme berechnet und lässt eine Vorhersage über das Mortalitätsrisiko des Patienten zu. Der Wert erfasst sieben verschiedene Risikofaktoren, wie zum Beispiel das Alter des Patienten, das Vorhandensein einer malignen Erkrankung, den Anteil der betroffenen Hautfläche sowie verschiedene Blutwerte wie Serum-Harnstoff und -Bikarbonat. Je mehr Risikofaktoren zutreffen, umso höher ist die prognostizierte Sterblichkeit (siehe Kasten „Letalität des SJS/TEN“).
Der wichtigste Schritt der Therapie ist es, das auslösende Arzneimittel sofort abzusetzen [9]. Aber gerade bei Patienten, die multimedikamentös betreut werden, kann es sich mitunter schwierig gestalten, den Verursacher zu identifizieren. Es ist wichtig, ein komplettes Medikationsprofil des Patienten zu erstellen, welches auch Präparate aus der Selbstmedikation berücksichtigt und alle Arzneimittel einschließt, die der Patient in diesem Zeitraum eingenommen hat. Genau hingeschaut werden sollte bei Arzneimitteln, die in den vorangegangenen acht Wochen angesetzt wurden [9]. Alle Wirkstoffe, die generell mit einem Stevens-Johnson-Syndrom/toxischer epidermaler Nekrolyse assoziiert sind, sollten sofort abgesetzt werden. Ebenso sollten früher aufgetretene Arzneimittelunverträglichkeiten bei der Suche nach dem Auslöser berücksichtigt werden [9].
Letalität des SJS/TEN
Um eine vorausschauende Aussage zur zu erwartenden Mortalitätsrate bei Patienten mit einem Stevens-Johnson-Syndrom sowie der toxischen epidermalen Nekrolyse zu treffen, wird der sogenannte SCORTEN-Score anhand prognostischer Faktoren ermittelt (SCORTEN = The severity-of-illness score for TEN)
prognostische Faktoren
Alter > 40 Jahre
Puls > 120 / Minute
maligne Grunderkrankung
betroffene Körperfläche > 10%
Serum-Harnstoff > 10 mmol/l
Serum-Bikarbonat < 20 mmol/l
Serum-Glucose > 14 mmol/l
Anzahl zutreffender Faktoren | Letalität [%] |
|---|---|
0 bis 1 | 3,2 |
2 | 12,1 |
3 | 35,3 |
4 | 58,3 |
> 4 | > 90,0 |
Supportive Therapie
Die weiteren Therapiemaßnahmen sind allesamt unterstützend und richten sich nach den auftretenden Symptomen. Eine Leitlinie zur Therapie des SJS/TEN ist derzeit in Bearbeitung. Die nachfolgenden Empfehlungen orientieren sich daher an der britischen Leitlinie [9]. Abhängig von der Schwere der Reaktion sollten die Patienten, bei denen mehr als 10% der Körperoberfläche betroffen sind, in einem spezialisierten Zentrum (zum Beispiel einem Verbrennungszentrum) betreut werden [9]. Da neben der Haut auch weitere Organe involviert sein können, sollten die Patienten dort interdisziplinär von Intensivmedizinern, Dermatologen, Ophthalmologen und Urologen behandelt werden.
Die Patienten müssen isoliert werden, um das Risiko einer Infektion möglichst gering zu halten. Pflegendes Personal sollte besonders vorsichtig mit dem Patienten umgehen, um eine Ablösung der Epidermis so gering wie möglich zu halten. Um den Flüssigkeitsverlust über die freiliegende Dermis auszugleichen und einer Minderdurchblutung wichtiger Organe sowie einem möglichen Kreislaufzusammenbruch entgegenzuwirken, muss meist Elektrolytlösung intravenös zugeführt werden. Die Flüssigkeitsmenge kann dabei anhand der ausgeschiedenen Urinmenge (> 0,5 bis 1 ml/kg Körpergewicht/Stunde) angepasst werden.
Die gesunde Haut sowie betroffene Areale sollten regelmäßig mit sterilem Wasser bzw. Kochsalzlösung, gegebenenfalls mit Chlorhexidin gereinigt und mit lipophilen Salben bzw. Sprays gepflegt werden. Die Haut in besonders betroffenen Bereichen sollte zudem mittels Abstrichen auf eine bakterielle oder candidöse Infektion überprüft werden. Bezüglich der Wundversorgung gibt es verschiedene Herangehensweisen, jedoch ohne herausragende Evidenz für ein bestimmtes Verfahren [9]. Die abgelöste Epidermis kann beispielsweise als „Wundauflage“ auf der Dermis belassen werden. Allerdings steht dieses Verfahren im Verdacht, die Infektionsgefahr zu erhöhen. Die freiliegende Dermis kann darüber hinaus auch mit nicht-adhäsiven Wundauflagen (z. B. nanokristalline Gazen oder Petroleum-getränkte Gazen) geschützt werden. Blasen sollten punktiert werden, um den Inhalt zu entleeren [9]. Bei Zeichen einer mikrobiellen Infektion sollte unverzüglich eine entsprechende antibiotische Therapie eingeleitet werden. Eine präventive Gabe von Antibiotika ist nicht ratsam, da sie ein unkontrolliertes Wachstum von Candida-Pilzen auf der Haut nach sich ziehen kann. Verschlechtert sich der klinische Zustand des Patienten und versagt das konservative Wundmanagement, so kann auch ein operativer Eingriff in Erwägung gezogen werden, um z. B. infizierte Abschnitte zu entfernen. Schleimhautbeteiligungen können ebenso nur symptomatisch behandelt werden. Bei einer Beteiligung der Augen sind z. B. befeuchtende Augentropfen zu applizieren und täglich die Augen mit Kochsalzlösung zu reinigen, um Ablagerungen zu entfernen und Verklebungen zu verhindern [9].
Ein weiterer wichtiger Bestandteil der supportiven Behandlung des Stevens-Johnson-Syndroms ist eine adäquate analgetische Therapie [9]. Die Patienten leiden aufgrund der teils großflächig freiliegenden Dermis unter starken Schmerzen. Um ihnen in Ruhe weitgehende Schmerzfreiheit zu ermöglichen, orientiert sich die Analgesie am Stufenschema der WHO. Nichtsteroidale Antirheumatika sind allerdings zu meiden, da sie die unter Umständen bereits angegriffene Magen-Darm-Schleimhaut und Nieren zusätzlich schädigen könnten. Bei leichten Schmerzen sollte Paracetamol verwendet werden, welches bei ungenügender Analgesie durch niederpotente Opioide wie Tramadol ergänzt werden kann. Bei starken Schmerzen werden dann hochpotente Opioidanalgetika wie Morphin oder Fentanyl eingesetzt. Besonders während pflegerischen oder therapeutischen Maßnahmen an schwer betroffenen Patienten können die Schmerzen unerträglich werden. Diamorphin und Remifentanyl sind mögliche Therapeutika, um in diesen Fällen Linderung zu schaffen. In schwersten Fällen können die Patienten oft nur unter Vollnarkose behandelt werden [9].
Über diese unterstützenden Maßnahmen hinaus gibt es wenig Evidenz für eine mögliche systemische adjunktive Therapie. Da das Stevens-Johnson-Syndrom bzw. die toxische epidermale Nekrolyse nur selten auftritt, sind groß angelegte randomisierte, kontrollierte klinische Studien entsprechend schwierig durchführbar. Derzeit werden verschiedene immunmodulatorische Ansätze diskutiert. Eine Therapie mit Glucocorticoiden ist dabei schon länger im Gespräch. Einerseits ließen sich so bei einigen Patienten die Spiegel wichtiger krankheitsassoziierter Zytokine wie Interferon gamma und Interleukin 6 signifikant reduzieren [11]. Jedoch erhöht die Immunsuppression die Gefahr von Infektionen und einer möglichen Sepsis. Aufgrund der derzeit ungenügenden Datenlage können systemische Glucocorticoide daher nicht empfohlen werden [9].
Ähnliches gilt auch für das Immunsuppressivum Cyclosporin A sowie für einen immunmodulatorischen Ansatz mit intravenösen Immunglobulinen. Die uneinheitliche Datenlage für beide Therapien lässt keine eindeutige Empfehlung zu [9]. Eine der wenigen randomisierten, kontrollierten Interventionsstudien zur Therapie des Stevens-Johnson-Syndroms und der toxischen epidermalen Nekrolyse wurde mit Thalidomid durchgeführt [12]. Der Wirkstoff hemmt die Produktion des Tumornekrosefaktors alpha (TNF alpha), und es wird angenommen, dass so der Krankheitsverlauf günstig beeinflusst werden kann. Die Studie wurde allerdings abgebrochen, da Thalidomid entgegen der Erwartungen die Mortalität signifikant erhöhte. Weitere TNF-alpha-Antagonisten wie Etanercept erwiesen sich in Einzelfällen als vorteilhaft [13].
Langzeitfolgen
Im Laufe der Behandlung klingen durch das Absetzen des betreffenden Arzneimittels die Blasenbildung und Epidermisablösung ab, und die betroffenen Hautareale werden langsam wieder re-epitelialisiert. Die Zeit bis zur Heilung variiert je nach Schweregrad und Komplikationen von einigen Tagen bis zu einigen Wochen. Häufig sind Spätfolgen der Erkrankung zu beobachten [2]. Da beim Stevens-Johnson-Syndrom und der toxischen epidermalen Nekrolyse nur die Epidermis betroffen ist, bilden sich im Laufe der Heilung meist keine Narben. Jedoch werden oft Pigmentationsstörungen (sowohl Hypo- als auch Hyperpigmentierungen) sowie Fehlbildungen der Nägel beobachtet. Eine Beteiligung der Augen verursacht langfristig oft trockene Augen. In schweren Fällen treten Hornhautschäden bis hin zu einem Visusverlust auf. War die Mundschleimhaut beteiligt, leiden Betroffene langfristig oft unter Xerostomie. Patienten müssen nach ihrer Entlassung darüber unterrichtet werden, die auslösenden Wirkstoffe und gegebenenfalls verwandte Verbindungen in Zukunft unbedingt zu meiden. |
Literatur
[1] Stevens AM und Johnson FC. A new eruptive fever associated with stomatitis and ophthalmia. Am J Dis Child 1922;24:526-533
[2] Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis 2010;5:39
[3] Mockenhaupt M et al. Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis: Assessment of Medication Risks with Emphasis on Recently Marketed Drugs. The EuroSCAR-Study. J Invest Dermatol 2008;128:35-44
[4] Halevy S et al. Allopurinol is the most common cause of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol 2008;58:25-32
[5] Coleman EL et al. The life-threatening eruptions of immune checkpoint inhibitor therapy. Clin Dermatol 2020;38:94-104
[6] Ramien M, Goldman JL. Pediatric SJS-TEN: Where are we now? F1000Res 2020:13;9:F1000 Faculty Rev-982
[7] Sharma AN et al. Stevens-Johnson syndrome and toxic epidermal necrolysis in pregnant patients: A systematic review. Int J Womens Dermatol 2020;6:239-247
[8] Abe R. Immunological response in Stevens-Johnson syndrome and toxic epidermal necrolysis. J Dermatol 2015;42:42-48
[9] Creamer D et al. UK guidelines for the management of Stevens–Johnson syndrome/toxic epidermal necrolysis in adults 2016. J Plast Reconstr Aesthet Surg 2016;69:736-741
[10] Bastuji-Garin S et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol 2000;115:149-153
[11] Hirahara K et al. Methylprednisolone pulse therapy for Stevens-Johnson syndrome/toxic epidermal necrolysis: Clinical evaluation and analysis of biomarkers. J Am Acad Dermatol 2013;69:496-498
[12] Wolkenstein P et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet 1998;352:1586-1589
[13] Paradisi A et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol 2014;71:278-283
Autor
Dr. Tony Daubitz, Studium der Pharmazie an der Universität Leipzig; Diplomarbeit in Basel an der Hochschule für Life Sciences der Fachhochschule Nordwestschweiz (FHNW) zu antientzündlichen Eigenschaften von Bambusextrakten; Promotion am Max-Delbrück-Centrum für Molekulare Medizin in Berlin zur Pharmakologie von Anionenkanälen



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.