













- DAZ.online
- DAZ / AZ
- DAZ 16/2021
- Zehn Jahre AMNOG
Foto: YK – stock.adobe.com
Politik
Zehn Jahre AMNOG
Gute Noten für ein lernendes System
Mit dem Gesetz zur Neuordnung des Arzneimittelmarktes in der gesetzlichen Krankenversicherung (AMNOG) wurde im Jahr 2011 die letzte Lücke der Reglementierung der Arzneimittelpreise geschlossen. Zu diesem Zeitpunkt war Deutschland das einzige verbliebene Land in der Europäischen Union, das im Segment der patentgeschützten Arzneimittel keinerlei Preisregulierung vorgenommen hatte. Die damit eingeführte „frühe“ Nutzenbewertung wird angewendet auf Zulassungen mit neuen Wirkstoffen und auch für neue Anwendungsgebiete bei bekannten Wirkstoffen. Bei Arzneimitteln für seltene Erkrankungen (Orphan Drugs) gilt der Zusatznutzen bis zu einer Jahresumsatzgrenze von 50 Millionen Euro als belegt. Der G-BA kann lediglich über das Ausmaß entscheiden. Auch Arzneimittel mit zu erwartenden geringen GKV-Ausgaben und Reserveantibiotika können auf Antrag davon freigestellt werden.
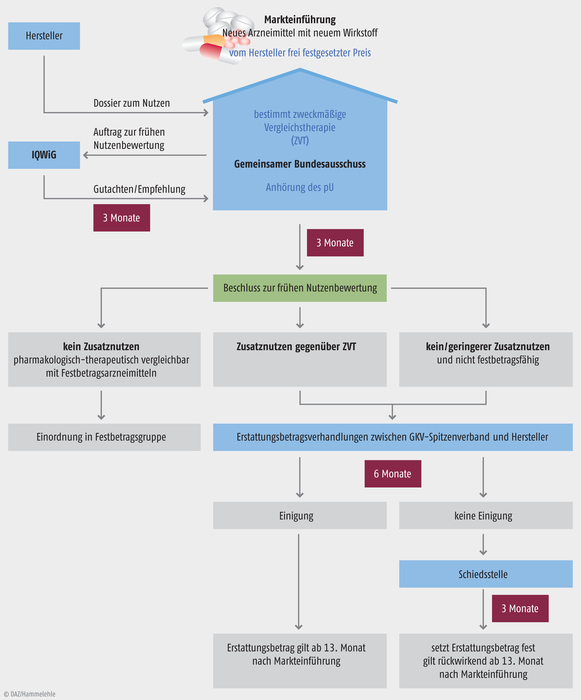
Abb. 1: Ablauf des AMNOG-Verfahrens
Wie läuft das AMNOG-Verfahren?
Die Nutzenbewertung wird als „frühe“ Nutzenbewertung bezeichnet, weil sie unmittelbar nach der Markteinführung in Deutschland durchgeführt wird. Das Prozedere hat sich seit der Einführung abgesehen von einigen Modifikationen im Grunde nicht geändert. Es besteht im Wesentlichen aus zwei Verfahrensabschnitten (Abb. 1):
- Nutzenbewertung durch IQWiG und G-BA: Zunächst bewertet der Gemeinsame Bundesausschuss (G-BA) innerhalb von drei Monaten nach Markteintritt eines neuen Arzneimittels, ob dieses gegenüber der zweckmäßigen Vergleichstherapie einen Zusatznutzen hat und wenn ja, wie groß dieser ist (§ 35a SGB V). Dazu müssen die Hersteller dem G-BA spätestens zum Zeitpunkt des erstmaligen Inverkehrbringens des Arzneimittels ein umfangreiches Dossier vorlegen. Für die wissenschaftliche Bewertung stützt sich der Bundesausschuss auf die Zuarbeit des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). Bei Arzneimitteln für seltene Erkrankungen (Orphan Drugs) ist die Abteilung Fachberatung Medizin des G-BA dafür zuständig.
- Preisverhandlungen zwischen dem GKV-Spitzenverband und Herstellern: Für Arzneimittel mit belegtem Zusatznutzen verhandeln die pharmazeutischen Unternehmen und der GKV-Spitzenverband danach innerhalb von sechs Monaten einen Erstattungsbetrag, der den „Wert“ des Arzneimittels widerspiegeln soll. Wenn die Bewertung keinen Zusatznutzen ergibt, wird das Arzneimittel nur noch mit dem für die jeweilige Arzneimittelgruppe bestimmten Festbetrag erstattet oder, falls keine passende vorhanden ist, mit einem Betrag, der nicht höher sein darf als für eine etablierte Vergleichstherapie. Das gesamte Verfahren inklusive Nutzenbewertung dauert regulär zwölf Monate. Spätestens ein Jahr nach Markteinführung eines Arzneimittels mit einem neuen Wirkstoff muss also klar sein, wie viel die Krankenkassen dafür bezahlen. Während dieses Zeitraums kann der pharmazeutische Unternehmer den Preis für sein neues Präparat allerdings noch frei bestimmen. Bis heute wird diskutiert, ob der finale Erstattungsbetrag nicht rückwirkend auch auf das erste Jahr des Inverkehrbringens angewendet werden müsste. Können sich der GKV-Spitzenverband und der Hersteller innerhalb eines Jahres nicht einigen, so entscheidet eine Schiedsstelle über den Erstattungsbetrag. Bis Ende 2020 gab es 46 solcher Schiedssprüche. Der verhandelte oder durch die Schiedsstelle festgelegte Erstattungsbetrag gilt nicht nur für die Versorgung im Rahmen der GKV, sondern auch für privat Versicherte und Selbstzahler.
Welche Nutzenkategorien gibt es?
Die möglichen Kategorien für die Bewertung eines Zusatznutzens sind in der Arzneimittel-Nutzenbewertungsverordnung (AM-NutzenV) sowie in der Verfahrensordnung des G-BA definiert. Diese orientiert sich wiederum an einem speziellen Methodenpapier (aktuelle Version 6.0 vom 05.11.2020) des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). Das Ausmaß des Zusatznutzens ist gestaffelt (Tab. 1). Für die ersten drei Kategorien wird zudem die Ergebnissicherheit für diesen Zusatznutzen bewertet. Dabei wird zwischen einem Anhaltspunkt, einem Hinweis oder einem Beleg für das Ausmaß des Zusatznutzens unterschieden.
Nutzenkategorien | Bedingungen |
|---|---|
erheblicher Zusatznutzen | nachhaltige und bisher nicht erreichte große Verbesserung des therapierelevanten Nutzens
|
beträchtlicher Zusatznutzen | bisher nicht erreichte deutliche Verbesserung des therapierelevanten Nutzens
|
geringer Zusatznutzen | bisher nicht erreichte moderate und nicht nur geringfügige Verbesserung des therapierelevanten Nutzens
|
nicht quantifizierbarer Zusatznutzen | wissenschaftliche Datengrundlage lässt keine Quantifizierung zu |
kein Zusatznutzen | kein Zusatznutzen belegt |
Quelle: G-BA (Orphan Drugs hier nicht ausgewiesen) | |
Der G-BA bemisst den Zusatznutzen ggf. differenziert bezogen auf Teilpopulationen, etwa wenn die Zulassung unterschiedliche Patientengruppen unterscheidet und wenn für diese verschiedene zweckmäßige Vergleichstherapien infrage kommen, oder wenn im Rahmen der zulassungsbegründenden Studien relevante Effektmodifikatoren beobachtet wurden. Deshalb wurden bereits ab dem ersten Nutzenbewertungsverfahren (Ticagrelor) Mischpreise vereinbart. Mischpreise gehören noch heute zu den bestimmenden AMNOG-Themen, denn es ist leicht vorstellbar, dass diese nicht einfach abzuleiten sind.
Knackpunkt zweckmäßige Vergleichstherapie
Ein besonderer Knackpunkt bei der Nutzenbewertung ist, dass der G-BA dafür in der Regel bereits vorhandene Therapieoptionen, das heißt eine zweckmäßige Vergleichstherapie (ZVT) heranzieht. Diese wird vom G-BA einseitig festgelegt. Erst durch eine Modifizierung der Vorschrift im Sozialgesetzbuch durch das Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV) von August 2019 besteht die Möglichkeit, die Fachkreise an der Festlegung zu beteiligen. Die ZVT muss dem aktuellen, anerkannten Stand des medizinischen Wissens entsprechen. Schwierig wird es dann, wenn neue Arzneimittel Therapielinien und -gebiete adressieren, zu denen auch die Kliniker keine Empfehlungen zu einer Standardbehandlung abgeben können.
Wie viele Verfahren wurden abgeschlossen?
Seit 2011 wurden bis Mitte April 2021 insgesamt 557 Nutzenbewertungsverfahren abgeschlossen, davon 150 für Orphan Drugs. Das Therapiegebiet mit den weitaus meisten Innovationen sind die onkologischen Erkrankungen (228 Verfahren), gefolgt von den Stoffwechselkrankheiten (105), Infektionskrankheiten (50), Krankheiten des Nervensystems (36) und Krankheiten des Atmungssystems (25). Im letzten Jahr waren von den insgesamt 87 abgeschlossenen Verfahren 18 Erst- und zehn Neubewertungen. 49 betrafen ein neues Anwendungsgebiet und zehn Orphan Drugs (> 50 Millionen Euro Umsatz). Nach Angaben im DAK-AMNOG-Report 2020 lag der durchschnittliche Preisabschlag nach Erstbewertung über die Jahre 2011 bis 2019 bei etwas über 21%.
Wie viele mit Zusatznutzen?
Im Schnitt bekommen seit Jahren etwa 60% aller bewerteten neuen Wirkstoffe einen Zusatznutzen zugesprochen. Ein erheblicher Zusatznutzen wurde bisher nur in sieben Fällen bescheinigt. Bei fast 24% aller Wirkstoffe bewertete der Bundesausschuss den Zusatznutzen als beträchtlich, bei rund 18% als gering und bei einem knappen Fünftel ließ er sich aufgrund der Studienlage nicht quantifizieren. Für etwa 40% der neuen Wirkstoffe wurde kein Zusatznutzen gesehen. Der Anteil ist über die Jahre hinweg in etwa konstant geblieben.
Über die Indikationsgebiete hinweg zeigen sich allerdings deutliche Unterschiede. Nach dem DAK-AMNOG-Report 2020 wurde (von 2011 bis Ende 2019) bei 69% der Bewertungen im Bereich Onkologika ein Zusatznutzen festgestellt (laut dem Verband Forschender Arzneimittelhersteller vfa sind es bis Mitte März 2021 sogar 79%), gegenüber 50% im Bereich Muskel-Skelett-System und lediglich 17% im Indikationsbereich Augenerkrankungen.
„Welche Schulnote geben Sie dem AMNOG?“
Im Rahmen des DAK-AMNOG-Reports 2020 wurde eine (nicht-repräsentative) Online-Befragung von 45 Experten von Krankenkassen(-verbänden), Kassenärztlichen Vereinigungen, Industrie(-verbänden), weiteren Institutionen der Selbstverwaltung und der Politik durchgeführt. Hiernach geben mehr als 70% der Befragten dem AMNOG die Note „gut“ bzw. „sehr gut“. Rund ein Viertel der Teilnehmer bewertet es als „befriedigend“ und lediglich ein Befragter erteilte die Note „ausreichend“. Die Noten „mangelhaft“ und „ungenügend“ wurden nicht vergeben.
Erstmals attestierte der G-BA im Jahr 2015 im Rahmen der frühen Nutzenbewertung einen erheblichen Zusatznutzen, und zwar Propranolol (Hemangiol®) für die Behandlung von Säuglingen mit proliferativen infantilen Hämangiomen (Blutschwämmchen), die eine systemische Therapie erfordern.
Ein weiteres Beispiel für ein neues Arzneimittel mit einem erheblichen Zusatznutzen, das nicht aus der Onkologie kommt, ist eine neue Wirkstoffkombination mit dem Handelsnamen Kaftrio® mit einer Triple-Therapie bestehend aus den Korrektoren Tezacaftor und Elexacaftor sowie dem Potentiator Ivacaftor für die Behandlung von Mukoviszidose bei Patienten ab 12 Jahren. Der pharmazeutische Unternehmer hatte dem G-BA trotz des gesetzlichen Orphan-Drug-Privilegs für sein Arzneimittel gleich ein vollständiges Dossier vorgelegt.
Gründe für den fehlenden Zusatznutzen
In vielen Fällen legen die Hersteller für die Nutzenwertung zulassungsrelevante Wirksamkeitsstudien vor, die das Arzneimittel jedoch häufig mit Placebo und nicht mit relevanten Standardtherapien vergleichen. Fehlende oder unpassende Vergleichsstudien (z. B. abweichende ZVT) sind also ein wesentlicher Grund für das Urteil „kein Zusatznutzen“. Um den Unternehmen dabei zu helfen, die Studien besser auf die Anforderungen der arzneimittelrechtlichen und gleichzeitig auch der Nutzenbewertung zuzuschneiden, haben der G-BA, das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und das Paul-Ehrlich-Institut (PEI) im April 2016 eine Vereinbarung getroffen, wonach die Unternehmen im Vorfeld der Planung klinischer Studien gemeinsamen Beratungsgespräche in Anspruch nehmen können.
13,4 Milliarden Euro an Einsparungen
Laut Berechnungen des vfa addieren sich die Einsparungen durch das AMNOG seit 2011 auf 13,4 Milliarden Euro. Nach anfänglicher Enttäuschung verläuft deren Wachstum nahezu exponentiell. 2020 erreichten die Entlastungen durch Erstattungsbeträge nach IQVIA-Daten 3,9 Milliarden Euro. Wie die Autoren des DAK-AMNOG-Reports 2020 ausführen, sind nur die Jahrestherapiekosten bei Orphan Drugs von 2012 bis 2019 sehr stark angestiegen (von rund 100.000 auf ca. 350.000 Euro), während sie bei den Nicht-Orphans mit rund 50.000 Euro nahezu konstant blieben.
Viele Befürchtungen nicht bewahrheitet
Nach der Analyse im AMNOG-Report 2020 haben sich viele anfängliche Befürchtungen in Bezug auf das AMNOG-Verfahren in der Praxis nicht bewahrheitet. So habe sich die Problematik vermehrter Marktrücknahmen als Opt-Out mit wachsender Zahl an Verfahren weitestgehend relativiert. Auch Erwartungen, dass die Unternehmen die freie Preisgestaltung im ersten Jahr regelhaft und strategisch ausnutzen könnten, hätten sich nicht bestätigt, da diese vielfach erst langsam im Markt angekommen seien. Weitere strittige Punkte, wie etwa die Bewertung des Bestandsmarktes oder eine mögliche späte Nutzenbewertung hätten sich ebenfalls weitestgehend entschärft, stellen die Autoren fest. Denn der G-BA spreche seine Nutzenbewertungsbeschlüsse immer häufiger befristet aus. So seien bis Ende 2019 bereits fast die Hälfte aller Arzneimittel innerhalb des erstzugelassenen Anwendungsgebietes erneut durch den G-BA bewertet worden.
Detaillierte Informationen zum AMNOG-Verfahren finden sich beim
IQWiG: www.iqwig.de/index.html → Projekte
G-BA: www.g-ba.de/bewertungsverfahren/nutzenbewertung/
GKV-Spitzenverband: www.gkv-spitzenverband.de/startseite/startseite.jsp → Krankenversicherung → Arzneimittel → AMNOG-Verhandlungen
International hervorragender Ruf
Bei einer virtuellen Jubiläumsveranstaltung des Gemeinsamen Bundesausschusses (G-BA) am 19. März 2021 fiel die Bilanz über zehn Jahre AMNOG überwiegend positiv aus. Das Verfahren habe sich „absolut etabliert“, und genieße „international einen hervorragenden Ruf“, hieß es dort. Da es sich stetig auf neue Herausforderungen einstellen müsse, sei das Verfahren nur als „lernendes System denkbar“. Als eine besondere Herausforderung der Zukunft führen Experten die hochpreisigen Arzneimittel für neuartige Therapien (ATMP) an. Viele davon werden in beschleunigten Zulassungsverfahren zugelassen. Das heißt, dass zum Zeitpunkt der Zulassung nur sehr begrenzte Kenntnisse zu Wirksamkeit und Sicherheit vorliegen. Weitere Herausforderungen sind die stärkere Einbeziehung des Kriteriums „Lebensqualität der Patienten“ in die Bewertung und die bessere Nutzung von Real World Data.
Europäische Nutzenbewertung „ante portas“
Das AMNOG ist aber in Sachen Nutzenbewertung neuer Arzneimittel noch lange nicht das Ende der Fahnenstange. Auch auf europäischer Ebene ist diese unter dem Begriff „Health Technology Assessment (HTA)“ schon seit Längerem ein Thema. Innerhalb Europas gibt es große Unterschiede, wann, wie und durch wen Nutzenbewertungen erstellt werden. Um Doppelarbeit zu vermeiden und Inkonsistenzen unter den Ländern zu reduzieren und damit letztendlich den Zugang der EU-Bürger zu innovativen Arzneimitteln zu verbessern, will die EU ähnlich dem Modell der europaweiten Zulassung eine europäische Nutzenbewertung einführen.
Im Rahmen eines Projektes namens „European Network for Health Technology Assessment“ (EUnetHTA, www.eunethta.eu/) waren die zuständigen Behörden in der EU schon seit einigen Jahren auf freiwilliger Basis zusammengerückt, um Unterschiede in den Bewertungen abzubauen. Damit das Ganze auch rechtlich festgezurrt wird, legte die EU-Kommission Ende Januar 2018 einen Vorschlag für eine Verordnung über die Bewertung von Gesundheitstechnologien vor. Hiernach sollten klinische Bewertungen auf nationaler Ebene durch harmonisierte Nutzenbewertungen obsolet werden. Die Entscheidungen über Preis und Erstattung sollten dagegen allein Sache der Mitgliedstaaten bleiben. Der Vorschlag sorgte in der Folge für erhebliche Diskussionen und Kontroversen. Vor allem größere Mitgliedstaaten wehren sich mit Vehemenz gegen eine Verbindlichkeit der harmonisierten Bewertungen für nationale Nutzenentscheidungen. Schließlich bewege sich jede Nutzenwertung im Kontext der nationalen Systeme der Gesundheitsversorgung, die sich ganz erheblich unterscheiden und diese unmittelbar beeinflussen, so die Argumentation.
Trotzdem will die EU-Kommission ihr Vorhaben nun bald in trockene Tücher bringen. Die aktuelle Fassung des Verordnungsvorschlags, über den jetzt verhandelt werden soll, sieht unter anderem die Einrichtung einer Koordinierungsgruppe aus nationalen Gesundheitsbehörden vor. Diese soll an gemeinsamen klinischen Bewertungen und wissenschaftlichen Konsultationen zu Gesundheitstechnologien arbeiten. Die Ergebnisse sollen jedoch die vorgelegte Evidenz nur beschreibend darstellen. Deren Bewertung soll weiter den HTA-Agenturen der einzelnen Mitgliedstaaten vorbehalten sein. Es sieht also aktuell nach einer EU-HTA „light“ aus. |
Literatur
DAK. AMNOG-Report 2020. 10 Jahre AMNOG – Rückblick und Ausblick. Beiträge zur Gesundheitsökonomie und Versorgungsforschung (Band 32). September 2020. file:///C:/Users/HB/AppData/Local/Temp/report-2335144.pdf
G-BA. Seit 10 Jahren als lernendes System etabliert: Nutzenbewertung von neuen Arzneimitteln. Pressemitteilung vom 19. März 2021. https://www.g-ba.de/presse/pressemitteilungen-meldungen/945/
G-BA. Neue Arzneimitteltherapie bei Mukoviszidose: G-BA vergibt Bestnote. Pressemitteilung vom 18. Februar 2021. https://www.g-ba.de/presse/pressemitteilungen-meldungen/934/
G-BA. G-BA vergibt erstmals höchste Zusatznutzen-Kategorie. Pressemitteilung vom 19. Februar 2015. https://www.g-ba.de/presse/pressemitteilungen-meldungen/571/
Verordnung über die Nutzenbewertung von Arzneimitteln nach § 35a Absatz 1 SGB V für Erstattungsvereinbarungen nach § 130b SGB V (Arzneimittel-Nutzenbewertungsverordnung - AM-NutzenV) vom 28. Dezember 2010 (BGBl. I S. 2324), zuletzt geändert durch Artikel 13 des Gesetzes vom 9. August 2019 (BGBl. I S. 1202). https://www.g-ba.de/richtlinien/42/
IQWiG-Jahresbericht 2019. https://www.iqwig.de/printprodukte/iqwig_jahresbericht_2019.pdf?rev=181052
Proposal for a Regulation of the European Parliament and of the Council on health technology assessment and amending Directive 2011/24/EU-Partial mandate for negotiations with the European Parliament. 2018/0018(COD) vom 24. März 2021. https://www.consilium.europa.eu/media/48963/st07310-en21.pdf
Vorschlag für eine Verordnung des Europäischen Parlaments und des Rates über die Bewertung von Gesundheitstechnologien und zur Änderung der Richtlinie 2011/24/EU vom 31.01.2018, https://eur-lex.europa.eu/legalcontent/DE/TXT/?uri=CELEX%3A52018PC0051
Autorin
Dr. Helga Blasius ist Fachapothekerin für Arzneimittelinformation, Dipl.-Übersetzerin (Japanisch, Koreanisch) und regelmäßige Autorin der DAZ.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.