
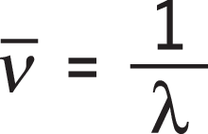
















- DAZ.online
- DAZ / AZ
- DAZ 17/2020
- Den Schwingungen auf der ...

Foto: skazar – stock.adobe.com
Analytik
Den Schwingungen auf der Spur
Wie funktionieren die IR- und NIR-Spektroskopie?
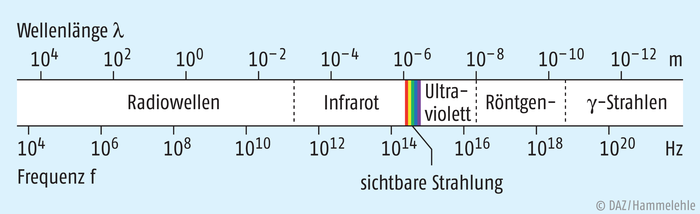
Innerhalb des breiten Spektrums der elektromagnetischen Wellenstrahlung kann unser Auge nur einen winzigen Bereich von Schwingungen mit Wellenlängen zwischen ca. 380 nm und 750 nm wahrnehmen. Hin zu kleineren Wellenlängen schließt sich der Bereich der ultravioletten (= UV-)Strahlung mit Wellenlängen zwischen 100 nm und 380 nm an. Nach oben grenzt das sichtbare Licht an den Bereich der Infrarot(= IR-)strahlung von 780 nm bis 1 mm (Abb. 1).
Abb. 1: Spektren-Bereiche der unterschiedlichen elektromagnetischen Wellen (nach [1])
Je kürzer die Wellenlänge bzw. je größer die Frequenz der unterschiedlichen Wellenstrahlung ist, desto energiereicher ist diese. Dies begründet auch die hohe Strahlenschädigung durch die sehr energiereiche Röntgen- bzw. γ-Strahlung. Dass es überhaupt zu Folgeerscheinungen der Einwirkung von elektromagnetischer Strahlung auf unseren Körper – wie Sonnenbrand durch UV-Strahlung oder Wärmeempfinden durch IR-Strahlung – kommt, lässt sich nur dadurch erklären, dass Wechselwirkungen zwischen der elektromagnetischen Strahlung und der biologischen Körpermaterie stattfinden. Diese beruhen vor allem auf einer Energieübertragung und damit Anregung von bestrahlter Materie.
Infrarotstrahlung
Die Infrarotstrahlung wird auch als Wärmestrahlung bezeichnet, weil sich Gegenstände unter ihrem Einfluss erwärmen. Die bedeutendste IR-Strahlungsquelle ist die Sonne. Je nach Wellenlänge unterscheidet man innerhalb des Spektralbereichs von Infrarotstrahlung unterschiedliche Arten von IR-Strahlung (Tab. 1).
Bezeichnung | Abkürzung | Wellenlänge in nm | |
|---|---|---|---|
nahes Infrarot | NIR | IR-A | 780 – 1400 |
IR-B | 1400 – 3000 | ||
mittleres Infrarot | MIR | IR-C | 3000 – 50.000 |
fernes Infrarot | FIR | 50.000 – 1.000.000 | |
Das Europäische Arzneibuch [3] grenzt die unterschiedlichen Bereiche der Infrarotstrahlung etwas anders ab als die DIN 5031 und ordnet dem mittleren Infrarotbereich eine Wellenlänge von 2500 bis 25.000 nm und dem nahen Infrarotbereich (NIR) eine Wellenlänge von 780 bis 2500 nm zu. Für die pharmazeutische Praxis ist diese Abweichung nicht von Bedeutung, da für die Identifizierung von pharmazeutisch relevanten Stoffen in der Apotheke der NIR-Bereich von 1000 bis 1900 nm völlig ausreichend ist [4]. Entsprechend finden sich bei den Allgemeinen Methoden des Europäischen Arzneibuches im Kapitel der „Methoden der Physik und Physikalischen Chemie“ die beiden Analysenmethoden „IR-Spektroskopie (2.2.24)“ und „NIR-Spektroskopie (2.2.40)“. Während die NIR-Spektroskopie im Ph.Eur. derzeit nur in 16 Monographien ausschließlich biologischer Produkte als eine zur Karl-Fischer-Methode bzw. zum Trocknungsverlust alternativ zugelassene Methode zur Bestimmung des Wassergehalts erwähnt wird, findet sich die IR-Spektroskopie in zahlreichen Arzneibuch-Monographien (vor allem organischer Substanzen) als Prüfung auf Identität. Dabei wird die IR-Spektroskopie teilweise als einzige Methode zur Identitätsbestimmung aufgeführt, in vielen Fällen als Alternative zu nasschemischen oder dünnschichtchromatografischen Prüfungen bzw. zu anderen physikalischen Methoden wie der Schmelztemperatur oder der Spezifischen Drehung.
Die IR-Spektroskopie
Die IR-Spektroskopie ist eine leistungsfähige Technik in der chemischen Analytik organischer Substanzen und wird zur quantitativen Bestimmung von bekannten Substanzen oder zur Strukturaufklärung unbekannter Substanzen genutzt. Sie ermöglicht direkte Aussagen über das Vorhandensein und ggf. die Konzentration IR-aktiver funktioneller Gruppen und basiert auf der Wechselwirkung von Infrarotstrahlung (hier konkret: mittlerer Infrarotstrahlung) mit Materie. Im Gegensatz zur UV-Vis-Spektroskopie, bei der durch Absorption von elektromagnetischer Wellenstrahlung im sichtbaren und ultravioletten Lichtspektrum Elektronen auf höhere Energieniveaus angeregt werden, führt die Interaktion von Materie mit Infrarotstrahlung zur Anregung von chemischen Bindungen. Durch Aufnahme konkreter Energiebeträge werden die Bindungen von organischen und z. T. anorganischen Molekülen zu Schwingungen angeregt. Dabei wird unterschieden zwischen
- 1. Valenzschwingungen (= Streckschwingungen), also Schwingungen in Richtung der Bindungsachse zweier Atome oder Molekülteile durch eine Dehnung oder Stauchung der Bindung und
- 2. Deformationsschwingungen (= Biege-/Beugeschwingungen), also Schwingungen unter der Deformation des Bindungswinkels.
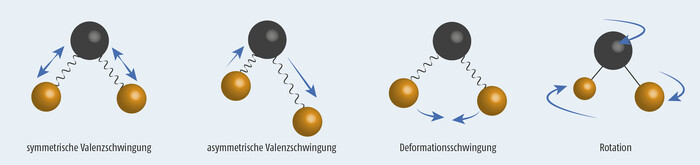
Abb. 2 zeigt die unterschiedlichen Grundschwingungen am Beispiel eines dreiatomigen gewinkelten Moleküls wie Wasser.
Wechselwirkung zwischen elektromagnetischer Strahlung und dem Molekül kann nur dann auftreten, wenn das Molekül entweder ein permanentes oder ein induzierbares Dipolmoment aufweist (IR-aktiv). Dies ist bei allen Schwingungen in Abb. 2 der Fall. Alle drei Grundschwingungen sind daher IR-aktiv. In Molekülen mit Schwingungen symmetrisch zum Symmetriezentrum treten keine Änderungen des Dipolmoments auf (IR-inaktiv). So absorbiert das symmetrische Wasserstoffmolekül H2 keine IR-Strahlung, die unsymmetrische Bindung im Chlorwasserstoffmolekül HCl hingegen ist schon IR-aktiv.
Foto: natros – stock.adobe.com
Abb. 2: Grundschwingungen am Beispiel des Wasser-Moleküls, nach [5]
Die zur Anregung von Molekülen notwendigen Energien (und damit Wellenlängen) sind abhängig von den schwingenden Massen und der Bindungsstärke und somit charakteristisch für die jeweiligen Bindungen. Dabei schwingen Atome und Moleküle umso langsamer, je größer deren Masse ist, und umso schneller, je stärker ihre Bindungen sind. Als Maß für die Energie der absorbierten Infrarotstrahlung wird in der Regel nicht die Wellenlänge λ angegeben, sondern die Wellenzahl ν_. Diese bezeichnet den Kehrwert der Wellenlänge, es gilt also:
Die Wellenzahl gibt die Anzahl der Schwingungen pro Längeneinheit an und besitzt die Einheit cm-1. Stärkere chemische Bindungen und Atome kleinerer Masse verursachen im IR-Spektrum Absorptionsmaxima bei großen Wellenzahlen (hoher Energie), große Massen hingegen verursachen IR-Absorptionsmaxima bei kleinen Wellenzahlen (niedriger Energie). Da Molekülschwingungen bestimmter Atomgruppen vor allem bei Wellenzahlen im Bereich von 4000 bis 1500 cm-1 besonders charakteristisch sind, eignet sich die IR-Spektroskopie zur Bestimmung der funktionellen Gruppen des untersuchten Moleküls. Im Gegensatz zu UV-Spektren, bei denen die Absorption gegen die Wellenlänge aufgetragen wird, wird in IR-Spektren die Transmission als Maß für die Durchlässigkeit der Anregungsstrahlung verwendet. Die Transmission wird nach oben zunehmend auf der vertikalen Achse gegen die Wellenzahl aufgetragen. Bereiche geringer Durchlässigkeit – also hoher Absorption – der IR-Strahlung ergeben eine Bande nach unten.
Ein typisches IR-Spektrum reicht von 4000 cm-1 bis 400 cm-1. Ab etwa 1500 cm-1 abwärts lassen sich die Transmissionen kaum mehr auf bestimmte Molekülgruppen zurückführen. Dies ist der sogenannte Fingerprint-Bereich. Anhand charakteristischer Absorption- bzw. Transmissionsbanden lassen sich bestimmte funktionelle Gruppen im untersuchten Molekül identifizieren. Tab. 2 enthält exemplarisch einige Molekülgruppen und die zugehörigen Bereiche der Transmissionsbanden.
Molekülgruppe | Transmissionsbande |
|---|---|
C-H | 3330 – 3270 cm-1 (Streckschwingung) |
C-H | 1400 cm-1 (Deformationsschwingung) |
C=C | 1680 – 1640 cm-1 |
C–––C | 2260 – 2100 cm-1 |
O-H | 3640 – 3160 cm-1 |
C=O | 1760 – 1670 cm-1 |
C–––N | 2260 – 2220 cm-1 |
N-H | 3500 – 3300 cm-1 |
NO2 | 1660 – 1500 cm-1 |
Meist lässt sich anhand eines IR-Spektrums schon auf den ersten Blick entscheiden, ob ein Alkohol, ein Amin oder ein Keton, eine aliphatische oder aromatische Verbindung vorliegt. Bei genauerer Betrachtung von Lage und Intensität einer Bande lassen sich jedoch noch sehr viel detailliertere Aussagen treffen, z. B. über den Substitutionstyp eines Aromaten oder über die Differenzierung zwischen einer freien Carbonsäure, einem Carbonsäureester und einem Carbonsäureamid. Begründet ist dies darin, dass „typische“ Werte von Transmissionsbanden durch die Umgebung der jeweiligen Bindungen etwas beeinflusst werden können. So sind Schwingungen eines Moleküls meistens nicht unabhängig voneinander, sondern miteinander gekoppelt. Vor allem auch elektronische bzw. mesomere Effekte können die Stärke einer Bindung und somit das IR-Absorptionsverhalten erheblich beeinflussen.
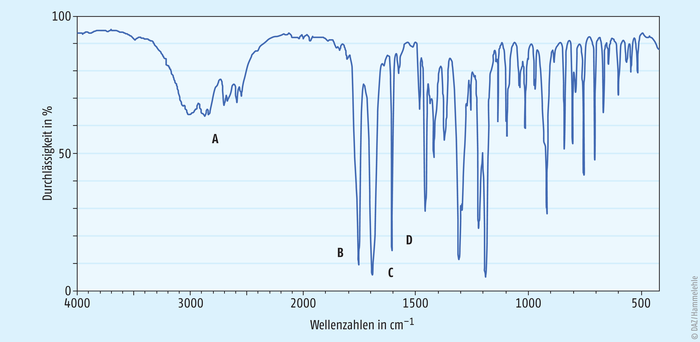
Abb. 3: IR-Spektrum von Acetylsalicylsäure [6]
Im Fingerprint-Bereich unterhalb 1500 cm-1 treten verschiedene Valenz- und Deformationsschwingungen vor allem von Einfachbindungen auf. Daneben zeigen sich hier auch sog. Gerüstschwingungen, die durch Schwingungen des gesamten Molekülgerüsts zustande kommen. Je größer und komplexer ein Molekül ist, desto vielfältiger sind die auftretenden Gerüstschwingungen. Solche Schwingungen sind nicht mehr charakteristisch für einzelne funktionelle Gruppen, sondern für das Molekül als Ganzes. Vor allem im Vergleich mit Referenzspektren aus Spektren-Datenbanken lässt sich oft eine eindeutige Zuordnung zu einer Substanz erreichen.
Abb. 3 zeigt exemplarisch ein IR-Spektrum von Acetylsalicylsäure. Darin zuordnen lassen sich:
- die O-H- und C-H-Valenzschwingungen bei 3300 bis 2500 cm-1 (A),
- die C=O-Valenzschwingung des Esters bei 1760 cm-1 (B),
- die C=O-Valenzschwingung der freien Carbonsäure bei 1700 cm-1 (C) sowie
- die C=C-Valenzschwingungen des aromatischen Rings bei 1600 cm-1 (D).
Techniken zur Aufnahme von IR-Spektren
Zur Aufnahme eines IR-Spektrums kann zwischen zwei hauptsächlichen Messarten unterschieden werden: der Transmission und der abgeschwächten Totalreflexion (attenuated total reflection = ATR).
Im Transmissions-Modus werden IR-durchlässige Proben von IR-Licht durchstrahlt und die Intensität der austretenden Strahlung mit der Intensität der einfallenden Strahlung ins Verhältnis (= Transmission) gesetzt. Da Glas nicht durchlässig für mittlere IR-Strahlung ist, können Substanzen nicht wie bei der UV-Vis-Spektroskopie in Küvetten in den Strahlengang gebracht werden. Transparent für MIR-Strahlung sind hingegen Salze wie z. B. Kaliumbromid. Lange Zeit wurden deshalb zur Messung eines IR-Spektrums von Festsubstanzen die Substanzproben mit Kaliumbromid vermengt und zu einer Tablette, dem sog. „KBr-Pressling“, verpresst. Diese sehr aufwendige und fehleranfällige Präparationstechnik ist heute weitgehend durch Reflexionstechniken verdrängt worden.
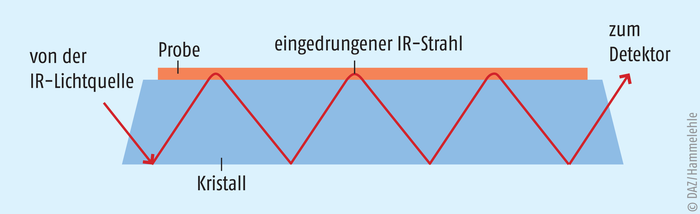
Im ATR-Modus dient ein Kristall aus Diamant als Messelement, auf dem die Probe aufgebracht wird. Das IR-Licht durchdringt den Kristall und tritt dabei mit dem Probenmaterial auf dem Kristall in Wechselwirkung. An der reflektierenden Grenzfläche kommt es theoretisch zu einer Totalreflexion. Praktisch kann die IR-Welle geringfügig (ca. 1 µm) in die Probe eindringen und dort ein Teil der Energie absorbiert werden. Die totalreflektierte Welle ist somit (je nach Zusammensetzung der Probe in unterschiedlichen Wellenlängen) abgeschwächt, wodurch ein Absorptionsspektrum erhalten wird. Abb. 4 zeigt das Prinzip der ATR-Messung.
Abb. 4: Prinzip der ATR-Messung (nach [7])
Das Europäische Arzneibuch [3] schreibt vor, dass zur Identitätsprüfung ein Vergleich des Spektrums der zu prüfenden Monographie-Substanz mit dem Spektrum einer Ph.Eur.-Referenzsubstanz (CRS) oder mit einem Ph.Eur.-Referenzspektrum durchgeführt werden muss. Ein gespeichertes Spektrum kann gemäß Ph.Eur. nur dann herangezogen werden, wenn „die Rückverfolgbarkeit zu einer aktuellen Charge einer CRS“ sichergestellt ist.
Beim Vergleich eines selbst gemessenen IR-Spektrums mit einem Referenz-Spektrum aus einer der zahlreichen kommerziellen oder frei verfügbaren Datenbanken oder Spektren-Bibliotheken wie z. B. der Spectral Database for Organic Compounds SDBS [6] oder der Spectrum Library von gasmet [8] muss stets beachtet werden, mit welcher Technik (Transmission oder ATR) das Literatur-Spektrum aufgenommen wurde, da sich mit unterschiedlichen Techniken aufgenommene Spektren ein und derselben Substanz teilweise ganz gravierend unterscheiden können.
Anwendung der IR-Spektroskopie
Vor allem für die Identifizierung von Substanzen ist die IR-Spektroskopie weit verbreitet. Sie kann auch zur quantitativen Bestimmung eingesetzt werden, wobei dann eine mathematische Beziehung zwischen der Intensität der gemessenen Absorptionsbanden und der Konzentration der untersuchten Substanz notwendig ist. Im pharmazeutischen Bereich wird die IR-Spektroskopie zur chemischen und physikalischen Analytik und Qualitätskontrolle von Wirkstoffen, Hilfsstoffen, Darreichungsformen und Verpackungsmaterialien eingesetzt. Im Rahmen einer physikalischen Analyse ermöglicht die IR-Spektroskopie beispielsweise die Bestimmung von Feststoffeigenschaften wie Polymorphie.
Limitiert wird der Einsatz zum einen durch die Tatsache, dass nicht alle Verbindungen IR-aktiv sind. Die Enantiomere einer Substanz können, zumindest bei der üblichen Verwendung von nicht linear polarisiertem IR-Licht, nicht unterschieden werden, da sie sich absolut IR-identisch verhalten. Da das IR-Verhalten von Festsubstanzen u. a. auch abhängig von deren Kristallform ist, können Bedingungen der Probenvorbereitung (wie Druck oder Lösungsmittel) die Kristallform von polymorphen Stoffen verändern. Außerdem kann für sehr heterogene Proben ein begrenztes Probenvolumen dazu führen, dass keine eindeutigen Ergebnisse erzielt werden.
Die NIR-Spektroskopie
Obwohl die „klassische“ IR-Spektroskopie mittels MIR-Strahlung in fast jeder Arzneibuch-Monographie organischer Substanzen als Methode der Identitätsbestimmung aufgeführt ist, ist in zahlreichen Apotheken kein MIR-Gerät zu finden. Ein wesentlicher Grund hierfür dürfte u. a. in den noch immer relativ hohen Anschaffungs- und Betriebskosten zu finden sein.
Eine kostengünstigere Alternative stellt die NIR-Spektroskopie dar, die mit nahem Infrarotlicht im Spektralbereich von 780 bis 2500 nm arbeitet. In diesem Wellenlängenbereich werden vor allem sogenannte Ober- und Kombinationsschwingungen der aus der (M)IR-Spektroskopie bekannten Grundschwingungen beobachtet. Oberschwingungen können aus der gleichzeitigen Aufnahme von mehreren Lichtquanten entstehen. Hauptsächlich treten Oberschwingungen auf, deren Grundschwingungen im Bereich zwischen 2500 bis 5000 nm liegen. Das sind im Wesentlichen Schwingungen von C-H-, N-H-, O-H- und S-H-Bindungen. Kombinationsschwingungen treten auf, wenn ein Lichtquant gleichzeitig zwei verschiedene Schwingungen anregt. Neben den molekularen Bindungsverhältnissen beeinflussen auch übermolelulare Eigenschaften wie die Anordnung im Kristallgitter das NIR-Verhalten. So können neben unterschiedlichen Kristallformen (Polymorphie) auch das Vorhandensein von Kristallwasser, Solvaten, Lösungsmittelrückständen oder auch die Partikelgröße oder die Schütt- bzw. Stampfdichte einen Einfluss auf das Aussehen des NIR-Spektrums haben. Auch äußere Faktoren wie die Umgebungstemperatur, die Luftfeuchtigkeit oder das Alter der Proben müssen in der praktischen Anwendung der NIR-Spektroskopie berücksichtigt werden [4].
Bei der Anregung zu Schwingungen höherer Ordnung wird im Gegensatz zur Anregung zu Grundschwingungen vergleichsweise wenig Energie absorbiert, woraus eine deutlich größere Eindringtiefe des NIR-Lichtes (im mm-Bereich) im Vergleich zum MIR-Licht (im µm-Bereich) resultiert. Eine Probenvorbehandlung kann dadurch meistens entfallen. So können Haufwerke von Festsubstanzen genauso gemessen werden wie halbfeste oder flüssige Zubereitungen. Die mit der Strahlenquelle und der Auswerteeinheit verbundene Messsonde kann bei Feststoffen direkt im Liefergefäß auf die Substanzoberfläche gehalten werden. Auch Flüssigkeiten können entweder in der Transmissionstechnik oder in der Reflexionstechnik unmittelbar mit dem IR-Licht bestrahlt werden. Da Glas weitgehend transparent für NIR-Strahlung ist, können im Gegensatz zur klassischen (M)IR-Spektroskopie NIR-Messungen durch die Gefäßwand von Probengefäßen aus herkömmlichen Glas durchgeführt werden.
Die Überlagerung der zusätzlichen Schwingungsmodi führt zu weitaus komplexeren NIR-Spektren mit einem größeren Informationsgehalt, deren Auswertung deutlich komplizierter als bei MIR-Spektren ist. Ein einfacher visueller Vergleich eines NIR-Spektrums mit einem Vergleichsspektrum lässt nur in Ausnahmefällen eine eindeutige Aussage zu. Vielmehr wird das Ergebnis computergestützt durch statistische Methoden, also durch chemometrischen Vergleich [9] mit den Daten einer validierten Referenzbibliothek verglichen. Unter Chemometrie versteht man die Anwendung mathematischer und statistischer Methoden zur Gewinnung von Informationen aus chemischen und spektroskopischen Daten.
Das Ph.Eur. fordert hinsichtlich der Erstellung einer Referenzbibliothek: „Die Spektren einer geeigneten Anzahl repräsentativer Proben der Substanz mit bekannten rückverfolgbaren Identitäten, bei denen die typische Variation der Probe (unter anderem die Festkörperzustände und die Partikelgröße) ausgeprägt sind, werden aufgenommen“ [10]. Idealerweise werden Proben aller Lieferanten einer Substanz vermessen. Auf diese Weise können Unterschiede z. B. in der Korngröße oder in der Kristallstruktur berücksichtigt werden. Je größer die Anzahl der Messungen unterschiedlicher Chargen von handelsüblichen Proben ist, desto valider kann die Entscheidung „Übereinstimmung“ oder „keine Übereinstimmung“ getroffen werden.
Anwendung der NIR-Spektroskopie
Nach A. Link können schätzungsweise 80% der apothekenrelevanten Ausgangsstoffe mittels NIR-Spektroskopie alleine oder in Kombination mit zusätzlichen Prüfungen identifiziert werden [11]. Die wesentlichen Vorteile sind zum einen in einer nichtdestruktiven Messung mit äußerst geringem Substanzverlust zu sehen. Die Messung kann in der Regel ohne Probenvorbereitung erfolgen und stellt damit eine massive Zeitersparnis im Vergleich zu klassischen Methoden wie z. B. der Schmelzpunktbestimmung oder der Dünnschichtchromatografie dar. Gerade im Vergleich zur Dünnschichtchromatografie oder zu nasschemischen Identitätsprüfungen kommt außerdem zum Tragen, dass bei der NIR-Spektroskopie quasi keine Chemikalien, also Gefahrstoffe, benötigt werden, womit aufwendige Anschaffungs-, Lagerungs- und Entsorgungskosten wegfallen. Wesentliche Nachteile sind die nicht geringen Investitionskosten und die natürlichen Limitierungen der NIR-Technologie. So sind anorganische Salze aufgrund ihrer IR-Inaktivität in der Regel nicht identifizierbar. Fette Öle sind bezüglich ihres Verhaltens gegenüber NIR meist so ähnlich, dass eine Differenzierung einzelner Öle oft nicht möglich ist. Und pflanzliche Drogen weisen einerseits innerhalb einer Droge häufig eine zu große Varianz auf, andererseits zeigen unterschiedliche Drogen manchmal sehr ähnliche NIR-Spektren.
In der Medizin wird die NIR-Strahlung aufgrund der großen Eindringtiefe beispielsweise bei Finger-Pulsoximetern zur Messung der Sauerstoffsättigung in kapillarem Blut genutzt. Deren Prinzip beruht auf der Tatsache, dass die Lichtabsorption von Hämoglobin (Hb) und mit Sauerstoff gesättigtem Hämoglobin (Hb-O2) unterschiedlich ist.
Gerätetypen
Prinzipiell lassen sich stationäre Labortisch-Geräte von akkubetriebenen mobilen Geräten unterscheiden.
Foto: apo-ident
Abb. 5: apo-ident® Analysensystem [13]
Bei den Labortisch-Geräten wie dem apo-ident® Analysensystem der Firma HiperScan GmbH (siehe Abb. 5) wird die Analysensubstanz mit einer Mindestfüllhöhe von 2 mm in ein Probenglas gefüllt und auf ein Messfenster aufgestellt. Nach Knopfdruck erfolgt die Messung im Spektralbereich 1000 bis 1900 nm bei einer spektralen Auflösung von 10 nm. Bei Flüssigkeiten und halbfesten Zubereitungen wird ein Stempel auf die Probensubstanz aufgesetzt, der das transmittierte IR-Licht wieder zurückspiegelt, sodass in einer Kombination aus Transmission und Reflexion gemessen wird. Außerdem sorgt der Stempel mit Abstandshaltern für eine ausreichende und definierte Schichtdicke. Die Software erstellt sowohl ein Prüfprotokoll als auch ein Qualitätskontroll-Etikett für das Standgefäß. Die Datenbank umfasst über 1100 vollständig validierte Ausgangsstoffe und kann im Internet eingesehen werden [12]. Die Software erlaubt entweder die gezielte Eingabe einer Eingangsvermutung zur Bestätigung oder Ablehnung oder bei unbekannten Substanzen die Anzeige der Rangliste mit der identifizierten Referenz auf Platz 1.
Foto: Apotec
Abb. 6: apotec® NIR-Gerät [14]
Bei dem apotec® System der Sentronic GmbH (siehe Abb. 6) handelt es sich um ein mobiles NIR-Spektrometer im Spektralbereich 900 bis 1700 nm (spektrale Auflösung 7 nm). Dieses kann mit einem Akku für eine Laufzeit von fünf bis sechs Stunden PC-unabhängig betrieben werden. Somit lässt sich das Gerät auch flexibel in einem Apothekenverbund bzw. in einer Filialapotheke nutzen. Der Messkopf mit einem kratzfesten Saphir-Fenster wird direkt in das Liefer- oder Standgefäß von Festsubstanzen bei einer Mindestschichtdicke von 5 mm eingetaucht, wodurch es zu annähernd keinem Substanzverlust kommt. Prinzipiell ist zwar auch eine Messung durch klare Glasgefäße möglich, diese wird jedoch vom Hersteller nicht empfohlen. Flüssige und halbfeste Substanzen werden in einer mitgelieferten Keramikschale bei einer Schichtdicke von 1 bis 2 mm vermessen. Im Gegensatz zum apo-ident® Analysensystem erfolgt hier die Messung „blind“, es muss also keine Vorgabe der zu analysierenden Substanz erfolgen. Das Messprotokoll wird zunächst im Gerät auf einer eingebauten microSD-Karte als PDF-Datei gespeichert. Über eine USB-Schnittstelle oder über WLAN findet die Sicherung der Messdaten und Prüfprotokolle auf einem PC bzw. das Aufspielen von Software-Updates statt. Eine integrierte Kamera erlaubt die gleichzeitige Dokumentation des Analysenzertifikats der Probe.
Die im Gerät hinterlegte Referenzdatenbank umfasst aktuell rund 1000 Substanzen, von denen jedoch nur ca. die Hälfte validiert ist. Nicht validierte Ergebnisse sind i. d. R. durch eine zu geringe Anzahl an hinterlegten Spektren von Referenzsubstanzen bedingt. Da nach dem Europäischen Arzneibuch [10] eine qualitative Analyse mittels NIR-Spektroskopie nur bei Vorhandensein eines validierten Verfahrens (und damit eines Vergleichs mit validierten Referenzspektren) zulässig ist, ist in diesen Fällen die Identifizierung und Charakterisierung von Ausgangsstoffen nicht anerkannt und es muss diese durch alternative Methoden erfolgen.
Beide Geräte müssen – auch nach einer Vorgabe des Ph.Eur. – regelmäßig kalibriert werden. Dies erfolgt z. B. mit einem Seltene-Erden-Standard entweder regelmäßig automatisch (bei apo-ident®) oder manuell durch den Nutzer (bei apotec®).
Die rechtliche Situation in der Apotheke
Prinzipiell handelt es sich bei der NIR-Spektroskopie um eine im Europäischen Arzneibuch unter dem Punkt 2.2.40 verankerte Methode und somit eine anerkannte pharmazeutische Regel, nach der Substanzen in der Apotheke geprüft werden können. Ausdrücklich aufgeführt ist sie im Ph.Eur. 9.8 jedoch nur zur Bestimmung des Wassergehalts in 16 Monographien. Zur Identitätsprüfung zahlreicher organischer Monographie-Substanzen ist ausnahmslos die „klassische“ IR-Spektroskopie unter Verwendung von mittlerem IR-Licht genannt. Andererseits lässt die Apothekenbetriebsordnung in § 6 Abs. 1 ausdrücklich den Einsatz alternativer Prüfmethoden zu, solange diese zu den gleichen Ergebnissen wie die herkömmlichen Arzneibuch-Methoden führen.
Die Arbeitsgemeinschaft der Pharmazieräte Deutschlands (APD) hat bereits in ihrer Jahrestagung 2013 die Identitätsprüfung mit Nahinfrarot-Spektroskopie in der Apotheke anerkannt. Im Oktober 2014 wurde dieser Beschluss erneuert und mit folgender Resolution am 1. Oktober 2014 konkretisiert: „Die Verwendung von Nahinfrarot ist eine anerkannte Prüfmethode nach Ph.Eur. 8. Für die Verwendung von NIR-Geräten in der Apotheke zur Prüfung der Identität von Ausgangsstoffen ist eine ausreichende und nachweisbare Validierung des verwendeten Gerätes erforderlich. Entscheidend ist die Qualität der vom Hersteller des Gerätes hinterlegten Datenbank. Chargenspezifische Unterschiede bei gleichen Ausgangssubstanzen müssen, wenn vorhanden, dabei berücksichtigt werden. Die Ausrüstung eines Apothekenlabors zur Identitätsprüfung von Ausgangsstoffen nach § 6 und § 11 ApBetrO ausschließlich mit einem NIR-Gerät ist nicht ausreichend. Ergänzende Möglichkeiten zur Identitätsprüfung müssen in der Apotheke vorhanden sein. Die Identität z. B. von anorganischen Salzen, von fetten Ölen und davon abgeleiteten Zubereitungen, von TCM-Drogen und daraus hergestellten Granulaten kann mit NIR allein nicht mit ausreichender Sicherheit geprüft werden. Unter Berücksichtigung der oben genannten Punkte und bei nachgewiesener und anerkannter Validierung kann NIR eine geeignete Methode zur Identitätsprüfung bestimmter Ausgangssubstanzen in der Apotheke sein“ [15]. |
Literatur
[1] Harten U. Physik. Springer Vieweg, Berlin, Heidelberg, 7. Auflage 2017:278
[2] DIN 5031-7: Strahlungsphysik im optischen Bereich und Lichttechnik; Benennung der Wellenlängenbereiche. Beuth Verlag Berlin 1984
[3] Europäisches Arzneibuch Ph.Eur. 9.8. Deutscher Apotheker Verlag Stuttgart. Methode 2.2.24 IR-Spektroskopie
[4] Link A, Wätzig H. Gute NIR-Praxis. Wissenschaftliche Verlagsgesellschaft Stuttgart 2016
[5] http://www.chemgapedia.de/vsengine/tra/vsc/de/ch/3/anc/ir_raman_spektroskopie1.tra/Vlu/vsc/de/ch/3/anc/ir_spek/methoden.vlu/Page/vsc/de/ch/3/anc/ir_spek/schwspek/methoden/ir_spek/ir4_1_3/dipolgew_m21ht0500.vscml.html (abgerufen am 16.02.2020)
[6] Spectral Database for Organic Compounds, SDBS: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi?lang=eng (abgerufen am 16.02.2020)
[7] Hug H. Instrumentelle Analytik. Verlag Europa-Lehrmittel Haan-Gruiten, 3. Auflage 2015:94
[8] Spectrum library von gasmet: https://www.gasmet.com/de/products/tools/spectrum-library/ (abgerufen am 16.02.2020)
[9] Europäisches Arzneibuch Ph.Eur. 9.8. Deutscher Apotheker Verlag Stuttgart. Allgemeine Texte 5.21 Chemometrische Methoden zur Auswertung analytischer Daten
[10] Europäisches Arzneibuch Ph.Eur. 9.8. Deutscher Apotheker Verlag Stuttgart. Methode 2.2.40 NIR-Spektroskopie
[11] Link A. NIR-Spektroskopie. Deutsche Apotheker Zeitung 2015;44:69
[12] https://hiperscan-download.de/software/public/available_substances_2019-05.pdf (abgerufen am 16.02.2020)
[13] Mit freundlicher Genehmigung der Firma HiperScan GmbH, Dresden
[14] Mit freundlicher Genehmigung der Firma WEPA Apothekenbedarf GmbH & Co. KG, Hillscheid
[15] https://www.pharmazierat.de/logicio/pmws/indexDOM.php?client_id=pharmazierat&page_id=resolutionen2014&lang_iso639=de (abgerufen am 16.02.2020)
Autor
Prof. Dr. Kurt Grillenberger, Pharmaziestudium und Promotion in Erlangen; Forschungstätigkeit in der Abteilung Nuklearmedizin des Universitätsklinikums Ulm; seit 1997 Dozent am Berufskolleg für PTA und an der Hochschule der Naturwissenschaftlich-technischen Akademie Prof. Dr. Grübler gGmbH; seit 2015 Rektor der nta Hochschule Isny; Lehrbeauftragter für Chemie an der Hochschule Kempten



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.