















- DAZ.online
- DAZ / AZ
- DAZ 11/2020
- Sartane
Repetitorium
Sartane
Beratungswissen zu Wirkungen, Nebenwirkungen und Interaktionen
Die Stoffklasse der „Sartane“ sind Angiotensin-II-Rezeptorantagonisten, d. h. sie blockieren im Renin-Angiotensin-Aldosteron-System (RAAS) die Wirkungen des Hormons Angiotensin II. Genauer blockieren sie mit hoher Selektivität einen der Subtypen dieses G-Protein-gekoppelten Rezeptors, den AT1-Rezeptor (AT1-Rezeptorantagonisten bzw. AT1R-Blocker). Sie sind Blockbuster in der Therapie von Hypertonie und Herzinsuffizienz. Seit mit Losartan der erste Wirkstoff dieser Klasse 1995 auf den Markt kam, haben fast alle großen Pharmafirmen Substanzen in dieser Wirkstoffklasse zugelassen. Es folgten 1996 das valinhaltige Valsartan, 1997 Eprosartan, Irbesartan und Candesartan. Welche 1998 mit Telmisartan, 2002 mit Olmesartan und 2012 mit Azilsartan ergänzt wurden. Die aus dem Patentschutz entlassenen Substanzen werden seither durch viele Generikafirmen angeboten [1]. Mit über 3300 verordneten definierten Tagesdosen liegt ihre Zahl noch unter der der ACE-Hemmer (z. B. Ramipril), jedoch sind die Zuwachsraten besonders bei Valsartan und Candesartan seit 2008 überproportional steigend [2]. Auch wenn die Mehrzahl der Wirkstoffe sich als Monopräparate auf dem Markt befinden, gibt es entsprechend dem Stufentherapieschema im Sinne einer verbesserten Compliance zahlreiche Kombinationspräparate mit Diuretikum (Hydrochlorothiazid) und/oder Calciumkanalantagonist (Amlodipin). Seit 2016 befindet sich daneben die fixe Kombination aus Valsartan und Sacubitril, dem ersten Vertreter der Neprilysin-Inhibitoren (Hemmstoff des Abbaus natriuretischer Peptide).
Indikation und Kontraindikation
Angiotensin-1-Rezeptorblocker (AT1-R-Blocker) zählen mit zur ersten Wahl bei der Therapie der arteriellen Hypertonie. Sie sind hier mit ACE-Hemmern gleichgestellt und werden bereits in der initialen Therapie als Mono- oder Kombinationspräparat mit Diuretikum oder Calciumkanalblocker empfohlen. Im Vergleich dazu sind Betablocker nur dann eine Option, wenn eine weitere Indikationsstellung oder Kontraindikation vorliegt [3]. Valsartan, Candesartan und Losartan sind darüber hinaus bei Herzinsuffizienz für den Fall indiziert, dass ACE-Hemmer nicht vertragen werden [4]. Einzelne Arzneimittel sind darüber hinaus in weiteren Indikationsgebieten zugelassen (Tab. 1). Zu beachten ist, dass die Zulassungen von Generika nicht in allen Fällen mit denen der Originalpräparate übereinstimmen.
Wirkstoff | Indikation |
|---|---|
Losartan | Hypertonie, chronische Herzinsuffizienz, Reduktion des Schlaganfallrisikos bei hypertonen Patienten, bei hypertonen Patienten mit Nephropathie13 |
Candesartan | Hypertonie, Hypertonie bei Kindern 6 – 18 Jahre, chronische Herzinsuffizienz14 |
Valsartan | Hypertonie, chronische Herzinsuffizienz, Nachbehandlung eines Herzinfarktes15 |
Irbesartan | Hypertonie, bei hypertonen Patienten mit Nephropathie16 |
Olmesartan | Hypertonie, Hypertonie bei Kindern 6 – 18 Jahre17 |
Telmisartan | Hypertonie, kardiovaskuläre Prävention18 |
Esprosartan | Hypertonie19 |
Kontraindiziert sind die AT1-R-Blocker bei Schwangeren, in der Stillzeit, bei Niereninsuffizienz oder Nierenarterienstenose sowie bei Hyperkaliämie [3] (Abb. 1). Eine Kombination mit anderen Stoffen, die in das RAAS eingreifen (z. B. ACE-Hemmer, Aliskiren (Renin-Inhibitor)), muss vermieden werden.
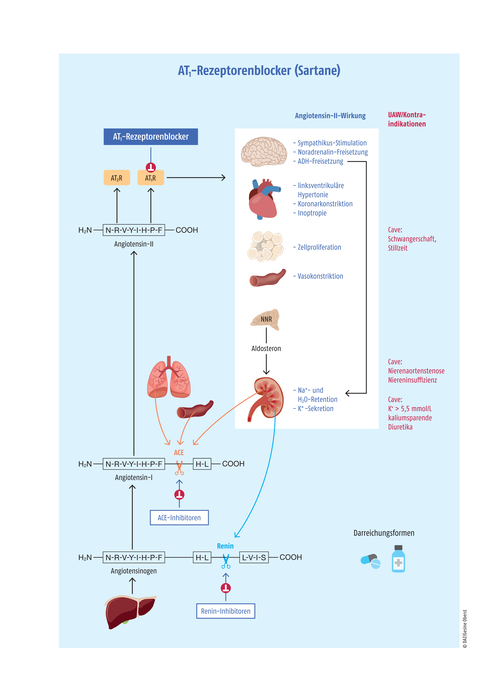
Abb. 1: Sartane Wirkungen, Nebenwirkungen und Kontraindikationen.
Wirkmechanismus und Nebenwirkungen
AT1-R-Blocker entfalten ihre Wirkung über einen Eingriff in das RAAS über eine Hemmung der G-Protein-gekoppelten AT1-Rezeptoren. Dieses System ist einer der zentralen Regelkreise des Körpers für Flüssigkeitsvolumen und Blutdruck. Renin stellt hierbei den Startpunkt der Kaskade dar.
Im juxtaglomerulären Apparat der Nieren werden vorlaufend Blutdruck, Blutvolumen und der Na-Ionenspiegel überwacht. Bei einem Abfall oder durch sympathische Aktivierung über β1-Rezeptoren wird das Enzym Renin aus seiner Vorläuferstufe Prorenin freigesetzt. Renin spaltet als Aspartyl-Protease aus Angiotensinogen in den Nieren das Angiotensin-I ab. Angiotensinogen selbst wird in der Leber gebildet und hat wie Renin keinen direkten Einfluss auf den Blutdruck. Das weitgehend inaktive Dekapeptid Angiotensin-I wird durch die zinkhaltige Peptidase Angiotensin-Converting-Enzyme (ACE, Metallo-Protease) zum Oktapeptid Angiotensin-II gespalten. ACE befindet sich hauptsächlich in der Lunge, der Leber und auf der Oberfläche von Endothelzellen in den Blutgefäßen, wodurch eine gewisse ACE-Aktivität im Plasma nachweisbar ist. Angiotensin-II nimmt als gefäßkontrahierendes Gewebshormon die Schlüsselposition im RAAS für die Aufrechterhaltung des Blutdrucks und des Wasserhaushalts über die glomeruläre Filtrationsrate ein. Über seine beiden Rezeptoren (AT1-R bzw. AT2-R) werden die regulatorischen Funktionen des RAAS vermittelt. Die Hauptrolle spielt hierbei der AT1-R, der AT2-R ist vor allem in der Fetalphase exprimiert und verliert im weiteren Verlauf an Bedeutung. Ihm wird eine vasodilatierende Funktion zugeschrieben. Ein weniger charakterisierter verwandter Insulin-regulierter AT4-R besitzt neben rezeptiven Arealen zusätzlich Enzymcharakteristika. Ein postulierter AT3-R konnte nicht bestätigt werden.
AT1-Rezeptoren werden am Herz, in den Nieren, im ZNS und im Gefäßsystem exprimiert. In den glatten Muskelzellen der Gefäße kommt es in der intrazellulären Signalkaskade über Gq/11-Kopplung zu einem erhöhten Ca2+-Spiegel. Dieser verursacht eine Vasokonstriktion und am Herzen eine positive Inotropie sowie eine Konstriktion der Koronarien. Im ZNS kommt es zur Stimulation des Sympathikus, die eine erhöhte Noradrenalin-Ausschüttung zur Folge hat. Über eine stimulierte ADH (Antidiuretisches Hormon, Vasopressin) -Freisetzung aus der Neurohypophyse (Hypophysenhinterlappen) kommt es zu einer erhöhten Wasserretention in den Nieren. Renal kommt es zur Ausschüttung von Aldosteron aus der Nebennierenrinde mit einer erhöhten Retention von Wasser und Na+. Angiotensin-II führt somit über ein erhöhtes Blutvolumen sowie einer starken Vasokonstriktion zu einer Blutdruckerhöhung. Durch eine selektive Blockade des AT1-R wird die Wirkung des Angiotensin-II verhindert (Abb. 2). Es wird diskutiert, dass durch AT1-R-Blockade das überschüssige Angiotensin-II AT2-Rezeptoren mit organprotektiven Effekten stimuliert (NO-Bildung, antihypertrophe Effekte auf glatte Muskelzellen und Myocard, Vasodilatation).
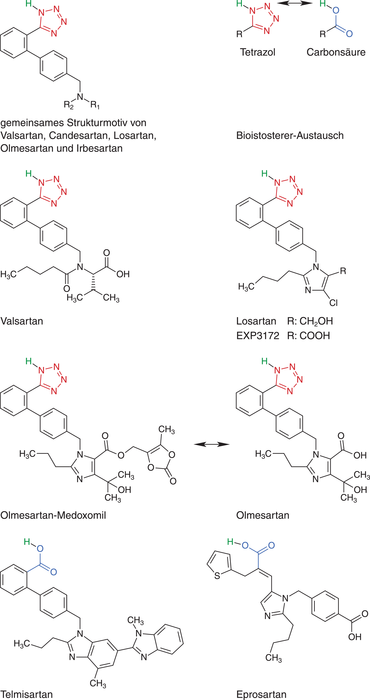
Abb. 2: Strukturübersicht einiger AT1-Rezeptorblocker, mit markiertem Tetrazolring (rot), Carbonsäure (blau) und saurem Wasserstoff (grün)
AT1-Rezeptorblocker werden meist gut bis sehr gut vertragen. Ihr unerwünschtes Arzneimittelwirkungsprofil (UAW) liegt auf dem Niveau von Placebo (Schwindel, Übelkeit, Hyperkaliämie, Hauterscheinungen, Allergien etc.). Typische Nebenwirkungen von ACE-Hemmern wie Bradykinin-induzierter Husten oder Angioödeme (Quincke-Ödem) treten seltener auf. Insbesondere wenn bei ACE-Hemmern Husten auftritt, kann ein Wechsel auf Sartane angeraten werden. Eine Kombination beider Wirkprinzipien verstärkt nicht deren Wirkungen, verstärkt aber mögliche schädliche Auswirkungen auf die Nieren. Bei schweren Leberschäden ist von einer Sartan-Therapie abzuraten.
Bei der Hochdruckbehandlung von Diabetikern werden Sartane mit organprotektiven Effekten besonders empfohlen, da sie beispielsweise einer diabetischen Nephropathie entgegenwirken. Neuere Studien legen einen zusätzlichen Benefit von Telmisartan mit zusätzlicher partial-agonistischer Wirkung an Peroxisom-Proliferator-aktivierten Rezeptor γ (PPARγ) oder Valsartan nahe, wo neben Klasseneffekten noch spezielle Substanzeffekte eine Rolle zu spielen scheinen.
Kontraindiziert sind AT1-R-Blocker bei Schwangeren und Stillenden. Im 1. Trimenon können Missbildungen entstehen, deren Ursachen noch nicht vollständig aufgeklärt sind. Im 2. und 3. Trimenon kann es zu einer Fetopathie kommen. Diese entsteht durch eine fetale Nierenfunktionsstörung und kann letal für den Fötus sein [5]. Das RAAS beeinflusst Zellwachstum und Gefäßneubildungen. Der Verdacht auf eine Erhöhung des Krebsrisikos durch spezielle Sartane konnte in umfangreichen Metaanalysen nicht bestätigt werden. Des Weiteren kontraindiziert sind AT1-R-Blocker bei Nierenaortenstenose und Niereninsuffizienz, da diese Patienten auf die vasokonstriktive AT1-R-vermittelte Angiotensin-II-Wirkung in den Nieren angewiesen sind. Eine Kombination mit Aliskiren bei Patienten mit Diabetes mellitus oder eingeschränkter Nierenfunktion ist eine absolute Kontraindikation.
Da die Substanzen den K+-Spiegel erhöhen können, sind sie bei einer bestehenden Hyperkaliämie ebenfalls kontraindiziert. Die Erhöhung des Kaliumspiegels ist eine Folge der verminderten Na+-Resorption in den Nieren, da hier der Austausch von Na+ gegen K+ blockiert wird. Von einer Kombination mit kaliumsparenden Diuretika wird daher ohne spezielle Bedarfslage ebenfalls abgeraten [1, 3, 6, 7]. Die Kaliumspiegel und Nierenwerte sollten regelmäßig kontrolliert werden. Wechselwirkungen mit Lithium-Präparaten oder nicht steroidalen Antirheumatika sind möglich.
Interessant ist, dass bei Menschen mit schwarzer Hautfarbe, die bereits weniger Renin im Blut haben, der blutdrucksenkende Effekt der Sartane geringer ausfällt.
Pharmakokinetik/Pharmakodynamik
Pharmakokinetisch betrachtet sind die verschiedenen AT1-Rezeptorenblocker eine zumeist homogene Gruppe. Die Halbwertszeiten der meisten Substanzen liegen zwischen 5 bis 10 h und der Plasmaspiegel-Maximalwert (tmax) wird nach 2 bis 4 h erreicht. Telmisartan zeigt hier eine abweichende Tendenz und flutet mit einem tmax von 0,5 bis 1 h schneller an, verbleibt dabei jedoch deutlich länger im Organismus (Plasmahalbwertszeit: 21 bis 38 h). Generell wird der erste Effekt auf den Blutdruck bei allen Substanzen nach zwei bis drei Stunden erzielt und stellt sich etwa nach zwei Wochen Behandlung ein. Der maximale blutdrucksenkende Effekt wird meist nach 6 h erreicht. Ein gesenkter Blutdruck kann durch alle AT1-R-Blocker mindestens 24 h nach Einnahme noch festgestellt werden.
Zahlreiche Sartane sind Prodrugs, die im Darm bei der Resorption oder von der Leber bioaktiviert werden. Candesartan wird als Candesartan-Cilexetil verabreicht, hierbei handelt es sich um ein Prodrug, welches in den aktiven Metabolit Candesartan gespalten wird [8, 9]. Auch Olmesartan ist als Olmesartan-medoxomil-Prodrug verfügbar. In beiden Fällen kommt es bei Aufnahme zu einer raschen Spaltung, die die aktive Wirkform (Candesartan bzw. Olmesartan) nach Esterspaltung und Hydrolyse freisetzt.
Aufgrund geringer Metabolisierungsraten der Wirkstoffe kommt es zu wenigen Interaktionen mit anderen Arzneistoffen oder Lebensmitteln. Losartan, bei welchem die Tetrazolgruppe metabolisch zur Carbonsäure umgewandelt wird (aktiver Metabolit EXP3174), wird verstärkt über CYP3A4 und CYP2C9 verstoffwechselt, sodass es zu Interaktionen mit Rifampicin und Fluconazol kommen kann.
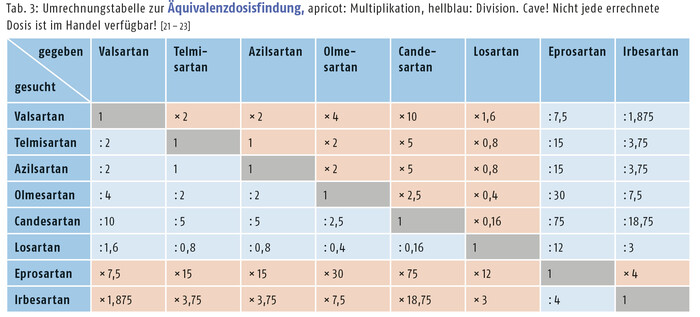
Eine Übersicht der Eigenschaften ist in Tabelle 2 dargestellt.
AT1-R-Blocker | Dosierung [mg] | Bioverfügbarkeit [%] | Wirkmaximum [h] | Halbwertszeit [h] | Plasmaeiweißbindung [%] |
|---|---|---|---|---|---|
Candesartan | 8 – 32 | ~ 15 (als Cilexetil) | 6 – 8 | 6 – 10 (kombiniert) | > 99 |
Valsartan | 80 – 320 | ~ 23 | 4 – 6 | 6 – 10 | 95 |
Losartan | 50 – 100 | ~ 33 | 6 | 2 6 – 9 (EXP3174) | > 99 |
Irbesartan | 150 – 300 | ~ 70 | 3 – 6 | 11 – 20 | 90 |
Olmesartan | 10 – 40 | ~ 40 (als Medoxomil) | 2 | 11 – 15 (kombiniert) | > 99 |
Telmisartan | 20 – 80 | ~ 50 | 3 – 9 | 21 – 38 h | > 99 |
Esprosartan | 600 – 800 | ~ 13 | 3 | 5 – 9 | 98 |
Azilsartan | 20 – 80 | 60 (als Medoxomil) | 1.5 – 3 | 11 | 99 |
Aktuelle Verunreinigungsproblematik
In die Schlagzeilen sind die AT1-R-Blocker in letzter Zeit vor allem durch eine weltweite Rückrufaktion gekommen. Bei Untersuchungen wurden potenziell krebserregende Substanzen in den Wirkstoffchargen gefunden. Zunächst war nur Valsartan betroffen, mittlerweile hat sich die Problematik jedoch auf fast alle weiteren tetrazolhaltigen Wirkstoffe ausgeweitet (Abb. 2; Valsartan, Candesartan, Losartan, Irbesartan und Olmesartan). Nicht betroffen von der Problematik scheinen Eprosartan und Telmisartan zu sein. Hier wurde statt des bioisosteren Tetrazolringes eine Carbonsäure-Gruppe in das Molekül integriert.
Bei den Verunreinigungen handelt es sich um Nitrosamine wie N-Nitrosodimethylamin (NDMA), N-Nitrosodiethylamin (NDEA). Die Verunreinigungen gehören zu der Klasse der Nitrosamine, welche als potenziell krebserregende Substanzen eingestuft sind [10, 11].
Nitrosamin-Problematik erklären
- Potenziell betroffene Sartane sind Valsartan, Losartan, Candesartan, Olmesartan und Irbesartan.
- Verunreinigte Chargen wurden aus dem Handel genommen.
- Neue Chargen werden auf Nitrosamine getestet.
- Die Erhöhung des Krebsrisikos wird als gering bewertet.
- Das Krebsrisiko steigt ca. um > 0,1% bei lebenslanger Einnahme eines verunreinigten Präparates.
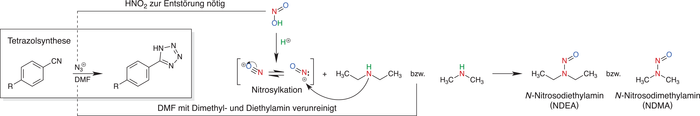
Die Verunreinigungen entstehen bei der Synthese des Tetrazolrings. Dabei entstehen sie nicht durch die eigentlichen Reaktanden, sondern durch Nebenreaktionen von Verunreinigungen aus dem Lösungsmittel und Hilfsreaktanden. Durch den Einsatz von reaktiverem und kostengünstigerem Natriumazid (NaN3), anstelle von Tributylinazid ((C4H9)3SnN3), ist es nötig geworden, die Reaktion mit HNO2 zu entstören (Abfangen von überschüssigem NaN3). Durch eine saure Umgebung kann aus der salpetrigen Säure ein Nitrosylkation gebildet werden. Dieses reagiert mit Verunreinigungen aus dem Lösungsmittel Dimethylformamid (DMF). Bei den Verunreinigungen handelt es sich um Dimethylamin bzw. Diethylamin. Diese Verunreinigungen werden zu den jeweiligen Nitrosaminen NDMA bzw. NDEA nitrosyliert. Der Mechanismus ist in Abbildung 3 dargestellt [12].
Abb. 3: Reaktionsprozesse zur Bildung von NDMA und NDEA
Obwohl die European Medicines Agency (EMA) das Risiko für die Patienten als gering einstuft, wurden im Sinne der Patientensicherheit entsprechende Produkte weltweit zurückgerufen und entsprechende Lieferengpässe generiert. Mit der Annahme, dass 100.000 Patienten täglich, über sechs Jahre lang ein verunreinigtes Präparat eingenommen haben, werden die zusätzlichen Krebserkrankungen auf 30 /100.000 über die Lebenszeit der 100.000 Patienten geschätzt. Wichtige Aspekte für die Patientenkommunikation sind im Kasten „Nitrosamin-Problematik erklären“ zusammengefasst [10].
so war das
- Sartane greifen in das Renin-Angiotensin-Aldosteron-System ein und antagonisieren die Wirkung von Angiotensin II, indem sie den AT1-Rezeptor blockieren.
- AT1-Rezeptoren befinden sich in unterschiedlichen Geweben, wie im Gehirn, der Lunge, dem Herz, den Gefäßen und den Nebennierenrinden.
- In Gefäßen kann die Angiotensin-II-Bindung an diese Rezeptoren zu einer Gefäßkontraktion und damit zur Blutdruckerhöhung führen, am Herzen zur Koronargefäßverengung und linksventrikulärer Hypertrophie
- Über die Blockade dieser Rezeptoren erklären sich die Wirkungen der Sartane und deren Einsatz bei arterieller Hypertonie und Herzinsuffizienz.
- Der Eingriff der Sartane in das Renin-Angiotensin-Aldosteron-System führt zu einer Einschränkung der Kalium-Sekretion, bei gleichzeitiger Behandlung mit Kalium-sparenden Diuretika droht eine Hyperkaliämie.
- Sartane sind gut verträglich, ein Bradykinin-induzierter Husten tritt unter ihnen seltener als unter ACE-Hemmern auf.
- Sartane sind in Schwangerschaft und Stillzeit kontraindiziert, ebenso bei Nierenaortenstenose und Niereninsuffizienz.
Fazit
Insgesamt stellt die Wirkstoffklasse der Sartane eine zuverlässige antihypertensive Therapieausrichtung dar. Aufgrund der gegenüber anderen Blutdruckmedikamenten erhöhten Tagestherapiekosten werden auch bei der Leitlinien-gerechten Therapie andere Optionen häufig bevorzugt. Darüber hinaus können Sartane indiziert sein bei chronischer Herzinsuffizienz und diabetischer Nephropathie. |
Literatur
[1] Steinhilber D, Schubert-Zsilavecz M, Roth H: Medizinische Chemie. (Deutscher Apotheker Verlag, 2010).
[2] Schwabe U, et al. Arzneiverordnungs-Report 2018. Onkologische Welt 09, (2019).
[3] Authors/Task Force Members: et al. 2018 ESC/ESH Guidelines for the managementof arterial hypertension. Europeam Heart journal 39, (2018).
[4] Bundesärztekammer (BÄK), Kassenärztliche Bundesvereinigung (KBV), A. der W. M. F. (AWMF). Nationale VersorgungsLeitlinie – Chronische Herzinsuffizienz – Langfassung, 2. Auflage. Version 3 2017. Äzq 147 (2017). doi:10.6101/AZQ/000405
[5] Embryotox – Arzneimittelsicherheit in Schwangerschaft und Stillzeit: Valsartan. Available at: https://www.embryotox.de/arzneimittel/details/valsartan/. (Accessed: 29th July 2019)
[6] Aktories K, Förstermann U, Hofmann F, Starke K: Allgemeine und spezielle Pharmakologie und Toxikologie. (Elsevier GmbH, 2017).
[7] Patel S, Rauf A, Khan H & Abu-Izneid T: Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 94,317-325 (2017).
[8] Song J C & White C M: Pharmacologic, Pharmacokinetic, and Therapeutic Differences Among Angiotensin II Receptor Antagonists. Pharmacotherapy 20,130-139(2000).
[9] Israili Z: Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J. Hum. Hypertens. 14,S73-S86(2000).
[10] Angiotensin-II-receptor antagonists (sartans) containing a tetrazole group | European Medicines Agency. Available at: https://www.ema.europa.eu/en/medicines/human/referrals/angiotensin-ii-receptor-antagonists-sartans-containing-tetrazole-group. (Accessed: 23rd July 2019)
[11] Charoo N A, Ali A A, Buha S K & Rahman Z: Lesson Learnt from Recall of Valsartan and Other Angiotensin II Receptor Blocker Drugs Containing NDMA and NDEA Impurities. AAPS PharmSciTech 20, 166 (2019).
[12] Snodin D J & Elder D P: Short commentary on NDMA (N-nitrosodimethylamine) contamination of valsartan products. Regul. Toxicol. Pharmacol. 103,325-329(2019).
[13] Fachinformation Losartan, Stand Januar 2019
[14] Fachinformation Candesartan Heumann, Stand April 2017
[15] Fachinformation Valsartan Heumann, Stand Dezember 2015
[16] Fachinformation Irbesartan Stada, Stand Juni 2016
[17] Fachinformation Olmetec, Stand März 2017
[18] Fachinformation Telmisartan Glenmark, Stand September 2016.
[19] Hypertonie, E., Erfahrung, K., Phosphokinase, K.– & Diuretika, K. Fachinformation Eprosartan Ratiopharm Stand März 2018.
[20] Äquivalenzdosen bei Umstellung: Wenn Valsartan-Patienten auf ein anderes Sartan umgestellt werden sollen. Available at: https://www.deutsche-apotheker-zeitung.de/news/artikel/2018/07/11/wenn-valsartan-patienten-auf-ein-anderes-sartan-umgestellt-werden-sollen. (Accessed: 6th January 2020)
[21] Giles T D, Oparil S, Silfani T N, Wang A & Walker J F: Comparison of increasing doses of olmesartan medoxomil, losartan potassium, and valsartan in patients with essential hypertension. J. Clin. Hypertens. (Greenwich). 9,187-195(2007).
[22] Sease J & Williams A M: Equivalent dosing of irbesartan, valsartan, and losartan identified through formulary switch at a Veterans Affairs medical center. Formulary2 43,14-20(8AD).
[23] Stalder M, Wegler R & Bornand D: Vergleichstabelle: Sartane. (2016). Available at: https://www.unispitalbasel.ch/fileadmin/unispitalbaselch/Bereiche/Querschnittsfunktionen/Spital-Pharmazie/vergleichstabelle_sartane.pdf
Autoren
Markus Falkenstein, Apotheker, Studium der Pharmazie an der Heinrich-Heine-Universität Düsseldorf; Praktisches Jahr an der Heinrich-Heine-Universität Düsseldorf und in der Apotheke am Schauspielhaus Bochum; Approbation 2017; seit 2017 Doktorand am Institut für Pharmazeutische und Medizinische Chemie der Heinrich-Heine-Universität Düsseldorf im Arbeitskreis von Prof. Dr. Dr. h. c. Holger Stark.
Prof. Dr. Holger Stark, Apotheker und Hochschullehrer; Studium der Pharmazie an der Freien Universität Berlin, Promotion 1991, Habilitation 1999, Professuren an der Goethe Universität Frankfurt 2000 (C3) und 2007 (W3), seit 2013 an der Heinrich-Heine-Universität Düsseldorf (W3) und seit 2015 Geschäftsführender Leiter der Pharmazie. Ehrendoktorwürde der Universität Nis, Serbien. Forschungsschwerpunkte sind in der Medizinischen Chemie Neurotransmitter wie Histamin, Dopamin und Glutamat sowie Lipidsignalling in Arachidon- und Sphingolipidkaskaden. Miterfinder des ersten selektiven Histamin-H3-Rezeptor-Antagonisten mit Marktzulassung, Pitolisant (Wakix®), als Orphan Drug gegen Narkolepsie. Sowohl für seine Forschungsarbeiten als auch für seine Lehre wurde er mehrfach ausgezeichnet.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.