





















- DAZ.online
- DAZ / AZ
- DAZ 20/2019
- „Unerwartet“

Foto: unpict – stock.adobe.com
Arzneimittelsicherheit
„Unerwartet“ verunreinigt
Eine kritische Betrachtung anlässlich einer neuen Meldung zu NDMA in Pioglitazon
Das Arzneibuch und andere Regularien
Das Europäische Arzneibuch und auch die United States Pharmacopoeia wollen mit den in den einzelnen Arzneistoff-Monographien beschriebenen Prüfungen auf Reinheit und Gehaltsbestimmungen („Tests“) die Qualität eines Wirkstoffes evaluieren und damit letztlich garantieren. Dabei wird ein umfangreiches Portfolio an standardisierten Methoden angewendet.
Die sogenannte Prüfung auf „verwandte Substanzen“, die zumeist mittels Hochleistungsflüssigkeits-chromatografischer Methoden durchgeführt wird, zielt bei organisch-chemischen Wirkstoffen weitestgehend auf organische, strukturverwandte Verunreinigungen ab. Sie entstammen dem Syntheseweg und sind klassischerweise Syntheseausgangs- und -nebenprodukte sowie Zersetzungsprodukte, in seltenen Fällen auch Verunreinigungen von Syntheseausgangsprodukten. Daraus ergibt sich, dass sich die beschriebenen Methoden auf einen Herstellungsweg, d. h. zumeist eine Syntheseroute, beziehen. Da die Syntheserouten einzelner Hersteller zu dem gleichen Wirkstoff zwar verschieden, aber dennoch ähnlich sein können, deckt die Testung auf „verwandte Substanzen“ in den jeweiligen Arzneibuchmonographien häufig mehrere Produktionswege ab. In manchen Monographien ist auch versucht worden, mit einer HPLC-Methode die Syntheseausgangs- und -nebenprodukte gänzlich unterschiedlicher Herstellungswege zu analysieren, was allerdings immer eine Herausforderung ist.
Es sei noch erwähnt, dass es Schwellen (= Grenzwerte) gibt, ab der eine Substanz, sprich Peak im Chromatogramm, zu identifizieren, zu berichten, zu spezifizieren und zu qualifizieren ist. Die jeweiligen Anforderungen sind an die Richtlinien der „International Conference on Harmonization“ (ICH), hier der Q3A(R2) „Impurities of New Drug Substances“, angelehnt [1]. Verunreinigungen, die qualifiziert werden müssen, sind im Arzneibuch spezifiziert, das heißt, sie haben ein Akzeptanzkriterium, das nicht weiter gefasst sein darf, als in den Zulassungsunterlagen, die von den zuständigen Behörden genehmigt wurden, beschrieben ist.
Die Richtlinie ICH Q3A(R2) befasst sich mit organischen Verunreinigungen (den bereits erwähnten verwandten Substanzen), anorganischen Verunreinigungen und Restlösemitteln, wobei bezüglich der Restlösemittel oder, allgemein gesagt, volatilen Substanzen auf die Richtlinie ICH Q3C(R7) „Impurities: Guideline for Residual Solvents“ [2] verwiesen wird. Die Menge des Restlösemittels aus Synthese und Kristallisation wird in der Regel mit der Dampfraum-Gaschromatografie bestimmt.
Komplettiert werden die ICH-Q-Richtlinien betreffend Verunreinigung durch die ICH-Q3D-Richtlinie [3]. Sie beschreibt die spezifische Prüfung auf Metallrückstände aus Katalysatoren oder Metallreagenzien bzw. auch aus verwendeten Synthese-Gerätschaften. Diese Rückstände können mittels Atomemissionsspektrometrie (AES), Atomabsorptionsspektrometrie (AAS), Massenspektrometrie mit induktiv gekoppeltem Plasma (ICP-MS), optische Emissionsspektrometrie mit induktiv gekoppeltem Plasma (ICP-OES) oder Röntgenfluoreszenzanalyse (RFA) bestimmt werden.
Mit anderen Worten: Entwickelt der Hersteller einen neuen Syntheseweg oder ändert er einen bestehenden, muss er gewährleisten, dass es mit den im Arzneibuch beschriebenen Methoden möglich ist, die möglichen neuen Verunreinigungen zu erfassen und damit zu begrenzen. Dies ist im Kapitel 5.10 des Europäischen Arzneibuches „Kontrolle von Verunreinigungen in Substanzen zur Pharmazeutischen Verwendung“ klar beschrieben und damit geregelt.
Mehr Nitrosamine als erwartet
Das hier summarisch dargestellte Regelwerk scheint relativ umfassend. Aber trotzdem ist man bei den Sartanen – wohl eher nur durch Zufall – auf die Nitrosamine gestoßen. Woran liegt das? Die Nitrosamine sind im engeren Sinne nicht strukturell verwandt mit den Sartanen. Damit werden sie auch mit Prüfung auf „verwandte Substanzen“ nicht erfasst. Wie kürzlich von uns in dieser Zeitschrift beschrieben [4], stammen die für die Bildung der Nitrosamine notwendigen Amine aus dem Lösungsmittel Dimethylformamid (DMF), das für die Synthese benutzt wird: DMF setzt leicht Dimethylamin frei und ist mit anderen Aminen, wie Diethyl- und Diisopropylamin, verunreinigt. Da bei der Synthese der Sartane, die einen Tetrazolylrest tragen, Nitrite zum Abfangen von überschüssigem Azid eingesetzt wurden und somit gleichzeitig mit den Aminen zugegen waren, haben sich die Nitrosamine N-Nitrosodimethylamin (NDMA), N-Nitrosodiethylamin (NDEA) und N-Nitrosodiisopropylamin (NDIA) gebildet. Kürzlich wurde auch noch von N-Nitroso-N-methyl-4-aminobuttersäure (NMBA) in Lorsartan berichtet. NMBA kann aus N-Methyl-4-aminobuttersäure in Gegenwart von Natriumnitrit (s.o.) in Analogie zu NDMA, NDEA und NDIA entstehen. N-Methyl-4-aminobuttersäure wiederum kann sehr leicht durch Hydrolyse des Lösungsmittels N-Methyl-2-pyrrolidon (NMP) entstehen. Die Patentliteratur [5] zeigt, dass NMP als Lösungsmittel bei der Synthese von Losartan zum Einsatz kommen kann.
In einem aktuellen Beitrag in der Deutschen Apotheker Zeitung [6] wurde beschrieben, dass NDMA in einigen Chargen des Antidiabetikums Pioglitazon des Herstellers Hetero Labs gefunden wurde. Die ersten Patente der Firma Takeda [7, 8] beschreiben „Prior-art“-Synthesen nach Abbildung 1. So lässt sich auch in diesem Fall die Entstehung von NDMA in Analogie zur Entstehung bei der Herstellung von Sartanen erklären: DMF kommt als Lösungsmittel im ersten Prozessschritt zum Einsatz. In einer Folgestufe erfolgt eine Diazotierung mit Natriumnitrit und einer Mineralsäure sowie nachgeschaltet eine Meerwein-Arylierung [9]. Man muss allerdings anmerken, dass bei anderen Synthesewegen auf die Diazotierung und somit den Einsatz von Natriumnitrit verzichtet werden kann, weil der Reaktionsweg nicht über eine Amin-Zwischenstufe, sondern über eine Aldehyd-Zwischenstufe erfolgt. Da Hetero Labs auch Sartane herstellen, kann NDMA auch durch Kreuzkontaminationen und/oder Recycling von Lösungsmitteln in Pioglitazon gelangt sein. In diesem Fall sollten alle Produkte von Hetero Labs vorsorglich untersucht werden.
Es versteht sich, dass für die Nitrosamine nun im Arzneibuch entsprechende Analytikmethoden beschrieben werden müssen, mit denen der Gehalt an Nitrosaminen gemessen werden kann. Empfindliche GC/MS- und LC/MS/MS-Methoden wurden in den letzten Monaten von verschiedenen „Official Medicines Control Laboratories“ (OMCLs) sowie einigen nationalen Arzneimittel-Kontrollaboratorien entwickelt. Sie sind in der Lage, Nitrosamine im sub-ppm-Bereich zu quantifizieren [4].
Die EMA hat kürzlich vorläufige Grenzwerte für NDMA und NDEA in den einzelnen Sartanen festgelegt gemäß der ICH-Richtlinie M7(R1) „Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk“. Sie richten sich nach der jeweiligen Dosis der Sartane [10]. Im ersten Schritt wird das Arzneibuch wahrscheinlich nur ein Produktionsstatement in die entsprechenden Monographien aufnehmen, das regelt, dass die Produktion so geartet sein muss, dass der Grenzwert für die Nitrosamine nicht überschritten wird. Im nächsten Schritt werden dann in den einzelnen Monographien Methoden oder eine allgemeine Methode für die Nitrosamin-Quantifizierung eingeführt werden.
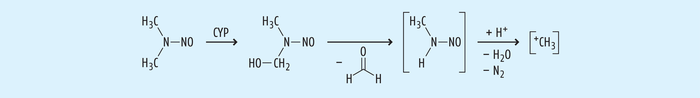
Gemein ist den Nitrosaminen, die wir auch in kleinen Mengen in Lebensmitteln wie Pökelfleisch oder alkoholischen Getränken wie Bier oder im Tabak und Tabakrauch finden, der Mechanismus der Toxifizierung, aus der die Gefährlichkeit hervorgeht (siehe Abb. 2): Cytochrom-P450-abhängige Monooxygenasen hydroxylieren α-Kohlenstoffe, dann wird chemisch Formaldehyd abgespalten und aus dem entstandenen Monoalkylnitrosamin wird unter Aufnahme eine Protons Wasser und N2 abgespalten. Zurück bleibt ein Alkylkation, das sowohl die DNA als auch Proteine leicht alkylieren kann [11].
Und was kann noch passieren?
Es stellt sich an dieser Stelle die Frage, welche anderen unerwarteten Verunreinigungen aus in der Synthese verwendeten Lösungsmitteln oder Reagenzien noch denkbar und möglich sind. Und wie kann man sie messen? Letzteres ist auf jeden Fall die berühmte Suche nach der Nadel im Heuhaufen. Im Folgenden wollen wir eine Auswahl von Verunreinigungen aufzeigen, die keine mit Wirkstoffen verwandten Verunreinigungen darstellen, sondern unerwartet sind und darüber hinaus einen „structural alert“ aufweisen, d. h. potenziell mutagen sind. Bei den im Folgenden aufgeführten Beispielen handelt es sich zum Teil um Wirkstoffsynthesen, bei denen der Wirkstoff nicht bis zur Marktreife entwickelt worden oder auch in Deutschland nicht zugelassen ist. Hier waren aber entsprechende Informationen aus der wissenschaftlichen Literatur recherchierbar. Sie stehen exemplarisch für eine Problematik, die für jede Wirkstoffsynthese zutreffen kann. Nur fehlen für aktuell zugelassene Wirkstoffe oftmals die relevanten Informationen, die nur den Zulassungsbehörden umfassend zugänglich sind.
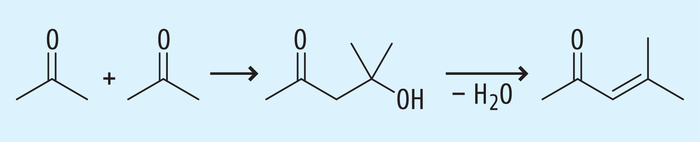
Beispiel 1a: Aceton und seine Aldolkondensationsprodukte. Beim Einsatz von Aceton als Lösungsmittel kann bei bestimmten Reaktionsbedingungen nach Abb. 3 eine Aldolkondensation stattfinden und über Diacetonalkohol Mesityloxid entstehen [12].
Folgt ein weiterer Reaktionsschritt in der Wirkstoffsynthese unter salzsauren Bedingungen, kann auch eine Reaktion des Mesityloxides mit Chlorwasserstoff stattfinden. Theoretisch gibt es zwei mögliche Regioisomere (siehe Abb. 4). Es kommt zur selektiven Bildung des höher substituierten Halogenids (Markovnikov-Regel), des 3-Cl-Derivats. Solche Alkylhalogenide werden zumeist metabolisch auf verschiedenen Wegen aktiviert, so dass sie ihre Toxizität entfalten können [11].
Beispiel 1b: Aceton bei Herstellung von Darreichungsformen. Aceton als Restlösungsmittel sowie die Aceton-Verunreinigungen wie Diacetonalkohol und Mesityloxid können auch bei der Herstellung der pharmazeutischen Darreichungsform eine große Rolle spielen. Sprühgetrocknete Dispersionen (Spray Dried Dispersions, SDD) sind einheitliche Mischungen, die aus einem bestimmten Verhältnis des Wirkstoffs in seiner amorphen Form und polymeren Hilfsstoffen bestehen. Sprühgetrocknete Dispersionen kommen oftmals dann zur Anwendung, wenn die Bioverfügbarkeit eines Wirkstoffs mit schlechter Löslichkeit im physiologischen pH-Wertbereich erhöht werden muss und eine Löslichkeitserhöhung durch Salzbildung nicht möglich ist. Leicht flüchtige Lösungsmittel wie Methanol, Tetrahydrofuran oder eben auch Aceton mit einem niedrigen Siedepunkt werden bevorzugt für die Sprühtrocknung verwendet, um den Wirkstoff und das Polymer zu lösen und um das Lösungsmittel beim Sprühvorgang ohne allzu hohe Energiezufuhr zu verdampfen. Bei Verwendung von Aceton sollte daher bei der Sprühtrocknung der Gehalt der Aceton-Zersetzungsprodukte Dicaetonalkohol und Mesityloxid kontrolliert werden [13].
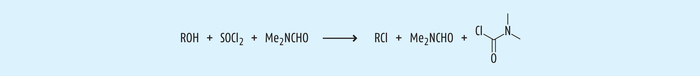
Beispiel 2: DMF und sein Vilsmeier-Produkt. Chlorierungen sind eine oft vorkommende Reaktion in der chemischen Synthese. Zur Chlorierung werden Chlorierungsmittel, häufig Thionylchlorid oder auch Phosphoroxychlorid, eingesetzt. Die Bildung von Säurechloriden aus Carbonsäuren durch Thionylchlorid wird katalysiert durch N,N-Dimethylformamid (DMF), welches auch als polares, aprotisches Lösungsmittel eingesetzt wird. Unter diesen Bedingungen kommt es neben der gewünschten Chlorierung des Wirkstoffs zur Bildung von N,N-Dimethylcarbamoylchlorid (DMCC) in einer Vilsmeier-Reaktion (nach Abb. 5, [14]). Seine große Toxizität rührt aus der Tatsache, dass DMCC mit jeglicher Hydroxylgruppe leicht Carbamate mit Proteinen bilden kann [11].
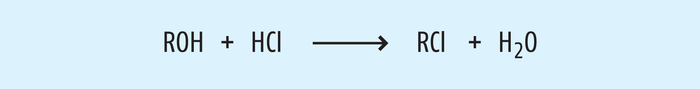
Beispiel 3: Chloralkane aus Alkoholen. Geeignete Salzformen von Wirkstoffen ermöglichen, Eigenschaften wie z. B. die Bioverfügbarkeit oder die Stabilität im Gegensatz zur freien Base zu optimieren. Eine breite Vielfalt stabiler Salze, wie z. B. Hydrochloride und Citrate, ist bekannt. Diese Salze sind auch in den Monographien der Arzneibücher beschrieben. Während des Salzbildungsschritts bei der Herstellung der Wirkstoffe werden häufig Alkohole (z. B. Methanol oder Isopropanol) verwendet, da sie die Fähigkeit haben, die freie Base sowie das Salz aufzulösen und einen kontrollierten Kristallisationsprozess zu ermöglichen. Folglich ist im letzten Syntheseschritt die Bildung von Methylchlorid bzw. Isopropylchlorids durch nukleophile Substitution (siehe Abb. 6) bei der HCl-Salz-Fällung möglich [15].
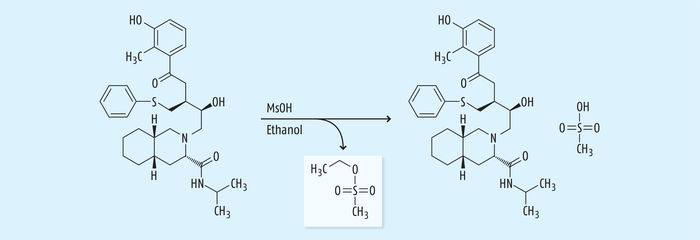
Beispiel 4: Bildung von genotoxischen Sulfonsäureestern. Eine Reihe von Wirkstoffen wird als Mesylat, Tosylat oder Besylat eingesetzt. Auch hier werden zur Salzbildung und Kristallisation ebenfalls sehr oft Alkohole eingesetzt, wobei sich als potenzielle Verunreinigungen die entsprechenden Sulfonsäureester aus der salzbildenden Sulfonsäure und dem Alkohol als Lösungsmittel bilden können, deren Vorhandensein als mögliche genotoxische Verunreinigungen im finalen Wirkstoff zu kontrollieren ist (Abb. 7) [16].
Ein historisch sehr wichtiges Beispiel, bei dem die Bildung einer solchen genotoxischen Verunreinigung einen großen Einfluss auf die globale Bereitstellung des Wirkstoffs hatte, war der Fall von Nelfinavir (Viracept®) zur Behandlung der humanen Immundefizienz-Virus(HIV)-Infektion. Im Juni 2007 führte eine Kontamination mit dem genotoxischen Sulfonsäureester Ethylmesylat zu einem weltweiten Rückruf des Arzneimittels (Abb. 8) [17]. Die Kontamination wurde intensiv untersucht, und es wurde festgestellt, dass die Hauptursache für eine Anreicherung der genotoxischen Verunreinigung beim letzten Herstellschritt lag. Bei diesem Schritt wird der Wirkstoff Nelfinavir als Mesylatsalz kristallisiert durch Zugabe von Methansulfonsäure zu einer Suspension von Nelfinavir in Ethanol. Die anschließende Sprühtrocknung des Mesylatsalzes aus der ethanolischen Lösung führte zu einer Anreicherung von Ethylmesylat, das bei der Freigabe nicht berücksichtigt worden ist [18].
Viele Patienten berichteten über einen strengen Geruch des Arzneimittels sowie über auftretende Nebenwirkungen wie Nausea nach Einnahme des Arzneimittels. Verantwortlich waren gravierende Qualitätsmängel im Rahmen des Herstellprozesses, weil die Kriterien der hohen Qualitätsanforderungen bei der Guten Herstellpraxis nicht umfassend beachtet worden sind. Hier wurde bei der Reinigung des Vorratstanks mit Ethanol vor Einfüllung der salzbildenden Säure nicht ausreichend getrocknet. Dieses Beispiel zeigt aber auch in eindrucksvoller Weise, dass die verschiedensten Prozesse im Rahmen der Herstellung für solche Reaktionen infrage kommen wie: Reinigungsprozesse zum Beispiel von Vorratstanks und Rohrleitungen, Standzeiten von Reagenzien und Lösungsmitteln, pH-Einstellung, Zugabegeschwindigkeit von Reagenzien während eines Reaktionsschrittes, verlängerte Reaktionszeiten oder zeitweise Überschreitung von kritischen Reaktionstemperaturen [18 – 21].
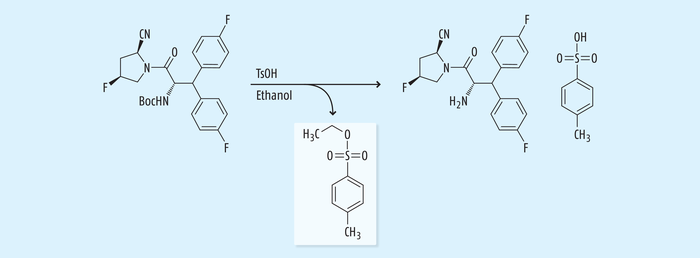
Dieses Beispiel steht für viele weitere Fälle, wo im salzbildenden Reinigungsschritt Sulfonsäuresalze in Verbindung mit Alkoholen als Lösungsmittel verwendet werden, wie durch ein weiteres Beispiel bei der finalen Salzbildung des Antidiabetikums Denagliptin-Tosylat gezeigt wird (Abb. 9). Bei der Zugabe von Tosylat in Ethanol als Lösungsmittel muss mit der Bildung von Ethyltosylat gerechnet werden [22].
Aufgrund der Toxizität der Alkylsulfonate, die auf ihren alkylierenden Eigenschaften beruht, werden diese im Europäischen Arzneibuch heute durch ein sogenanntes „Produktions-Statement“ begrenzt [23]. Der Hersteller muss durch den Produktionsprozess garantieren, dass kein Alkylsulfonat im Arzneistoff enthalten ist, wie z. B. in der Monographie Bromocriptinmesilat: „Bei der Entwicklung des Herstellungsverfahrens müssen die Grundsätze des Qualitätsmanagements unter Berücksichtigung der Qualität der Ausgangsmaterialien, der Leistungsfähigkeit des Herstellungsverfahrens und dessen Validierung angewendet werden.“ Unter den allgemeinen Methoden (PhEur Kap. 2.5.38 und 2.5.39) wird dann eine Dampfraum-GC/MS-Methode beschrieben, mit der der Hersteller im ppm-Bereich die Alkylsulfonate quantifizieren kann [24].
Beispiel 5: Bildung der genotoxischen Verunreinigung Acetamid bei Verwendung von Acetonitril als Lösungsmittel. Acetamid ist als Karzinogen bekannt (Kategorie I, belegt durch Tierstudien, Cardiogenicity Scope Factor (CSF) 0.07 mg/kg/day), daher ist eine mögliche Bildung als Nebenprodukt von besonderer Bedeutung während des Herstellprozesses. Zwar wird Acetamid nur selten direkt in der Wirkstoffsynthese verwendet, aber Derivate des Acetamids wie N-Bromoacetamid oder Trifluoracetamid werden häufiger als sogenannte Synthese-Bausteine (building blocks) verwendet. Diese Reagenzien neigen zur Bildung von Acetamid in Abhängigkeit von den angewandten Reaktionsbedingungen. Eine weitere Quelle für die Bildung von Acetamid ist das immer noch häufig verwendete Lösungsmittel Acetonitril. Es entsteht hier bei erhöhten Temperaturen unter sauren, aber auch basischen Bedingungen durch Hydrolyse [18].
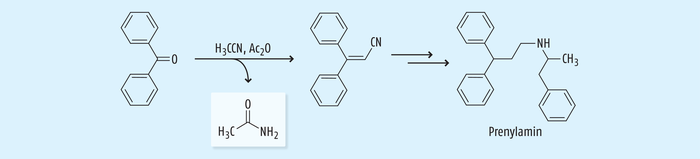
Acetonitril wird aber nicht nur als Lösungsmittel in der pharmazeutischen Industrie verwendet, sondern es findet auch Anwendung als Reagenz in der Wirkstoffsynthese. So kam zum Beispiel Acetonitril als Reagenz bei der Synthese von Prenylamin (Corontin®, Segontin®) zur Anwendung (Abb. 10), das zur Behandlung von Angina Pectoris eingesetzt wurde. Allerdings wurde der Wirkstoff bereits 1988 in USA, Kanada und UK wegen kardialer Arrhythmien vom Markt genommen [18, 25].
Zaurategrast ist ein Wirkstoff, der zur Behandlung der Multiplen Sklerose in der Entwicklung war, dessen klinisches Programm allerdings zwischenzeitlich eingestellt worden ist.Beim Herstellprozess wird in der letzten Stufe Acetonitril als Lösungsmittel unter sauren Bedingungen verwendet. Die Minimierung und Abreicherung von Acetamid war ein entscheidender Faktor während der Prozessoptimierung (Abb. 11). Hier wurde der Acetamidgehalt im Endprodukt durch eine mehrfache wässrige Aufarbeitung und eine finale Kristallisation zur Abreicherung (Purging) erfolgreich kontrolliert [26].
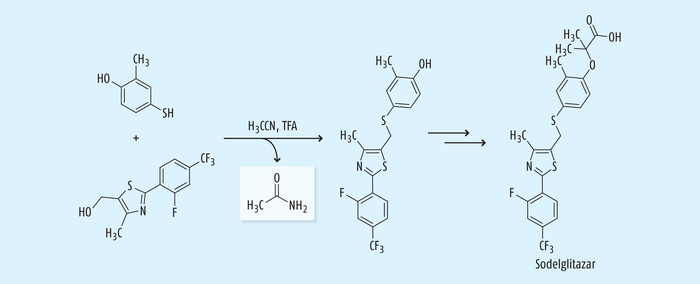
Sodelglitazar ist ein Agonist der Peroxisom-Proliferator-aktivierten Rezeptoren (PPAR), der zur Therapie des Diabetes mellitus Typ 2 gedacht war, dessen weitere Entwicklung wie die weiterer PPAR-Agonisten wegen Sicherheitsbedenken und fehlender Wirksamkeit eingestellt worden ist. Während der Synthese erfolgt eine S-Alkylierung in Acetonitril unter sauren Bedingungen, bei der Acetamid als Nebenprodukt aus Acetonitril (Abb. 12) gebildet wird [27].
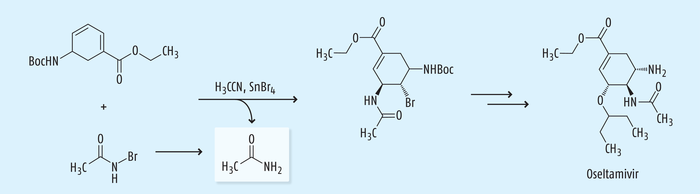
Während der Synthese des Wirkstoffs Oseltamivir zur Behandlung von Influenza wird eine Dien-Zwischenstufe zu einem Bromodiamid umgewandelt, wobei eine neuartige SnBr4-katalysierte Bromoacetamidierung mit N-Bromoacetamid in Acetonitril stattfindet (Abb. 13). Bei dieser Reaktion kann Acetamid sowohl aus dem Lösungsmittel wie aus dem Reagenz gebildet werden [28, 29].
Das Reagenz 2-Bromoacetamid kommt auch bei der Herstellung von Armodafinil,
in den USA zur Behandlung von Narkolepsie und Schlafstörungen zugelassen, zur Anwendung. Auch hier wird das Reagenz zur S-Alkylierung – hier allerdings unter basischen Bedingungen – eingesetzt (Abb. 14) [30, 31].
Insgesamt ist eine potenzielle Acetamid-Kontamination bei vielen Wirkstoffen durch den Einsatz von Acetamid-bildenden Reagenzien und Lösungsmitteln ein eher allgemeines Problem, das bei der Optimierung des Herstellprozesses so früh wie möglich zu berücksichtigen und die Abreicherung durch entsprechende Maßnahmen wie wässrige Aufarbeitungsschritte und mögliche Kristallisationen sicherzustellen ist. Diese Maßnahmen werden aber sicherlich die Gesamtausbeute bei der Herstellung reduzieren und damit die Kosten für den Wirkstoff in die Höhe treiben. Aber Qualität hat auch ihren Preis.
Beispiel 6: Dichlormethan als Alkylans. Es versteht sich, dass CH2Cl2 als Alkylhalogenid alkylierenden Charakter hat, auch wenn es weniger reaktiv ist als die verwandten Alkylbromide und -iodide. Da es als Lösungsmittel in einer Reaktion stets in extremem Überschuss vorliegt, muss mit einer Alkylierung von z. B. Aminen gerechnet werden. So wurde von Dondre et al. [32] die Bildung quartärer Ammoniumsalze des Typs R3N-CH2Cl von Chloroquin und Hydroxychloroquin berichtet, die offenbar in Mengen > 0,1% vom Wirkstoff vorliegen können und deshalb spezifiziert werden müssen. Die Ammoniumsalze selber sind wahrscheinlich nicht toxisch, sie können aber – wie in Atracuriumbesylat zu beobachten ist – eventuell einem Hofmann-Abbau unterliegen und damit R2NCH2Cl bilden, was alkylierend sein kann [33].
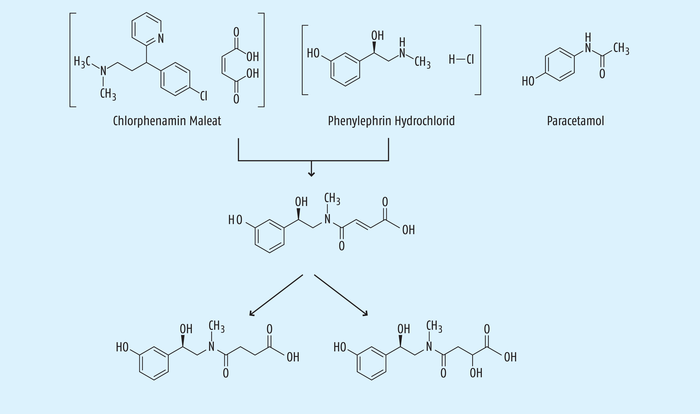
Beispiel 7: Die unerwartete Bildung von Verunreinigungen in der fertigen Arzneiform. Nicht nur bei der Herstellung von Wirkstoffen kann es zu einer unerwarteten Bildung von Verunreinigungen kommen, sondern auch bei fertigen Arzneiformen, die zum Teil schon seit vielen Jahren auf dem Markt sind. Es wurden Kombinationspräparate untersucht, die als Wirkstoffe Paracetamol, Phenylephrin-Hydrochlorid und Chlorpheniramin-Maleat enthalten und besonders auf dem US-amerikanischen Markt als OTC-Produkte vertrieben werden. Überraschenderweise kommt es unter Lagerbedingungen zu einer Veresterung des Maleations aus dem Chlorphenamin-Maleat mit dem Phenylephrin unter Bildung des entsprechenden Esters des Phenylephrins (Abb. 15). Nach erstmaliger Identifizierung einer solchen Verunreinigung konnte dann in der Folge festgestellt werden, dass die meisten Marktprodukte diese Verunreinigung enthielten, auf die aber zuvor niemals geprüft worden ist [34].
Schlussfolgerung
Eine detaillierte Bewertung der gesamten Wirkstoffsynthese beginnend bei den Startmaterialien unter Berücksichtigung der verwendeten Hilfsstoffe (Lösungsmittel und Reagenzien) sowie deren potenziellen Verunreinigungen über jeden einzelnen Syntheseschritt unter Berücksichtigung der Nebenreaktionen, Folgereaktionen und Umwandlungen von Verunreinigungen sowie Verschleppungen der resultierenden potenziellen Verunreinigungen bis hin zum finalen Wirkstoff ist essenziell. Das Ziel dieser Bewertung ist festzustellen, basierend auf dem Wissen der ablaufenden chemischen Reaktionen, welche Verunreinigungen potenziell im Wirkstoff enthalten sein können, ihr Gefährdungspotenzial abzuschätzen und geeignete Kontrollmaßnahmen (analytische Prüfungen) zu definieren.
Sicher kann man keine Qualität in einen Arzneistoff oder ein Arzneimittel hinein prüfen. Da uns aber immer wieder neue Verunreinigungen überraschen, die strukturell nicht mit dem Wirkstoff verwandt sind, sollte man neben der Arzneibuchanalytik auch unabhängig mit den Methoden der ungezielten Analyse, besser bekannt unter dem englischen Terminus „untargeted analysis“, wie LC/MS/MS gekoppelt mit chemometrischen Auswertungen, die Qualität eines Arzneistoffes überprüfen. Im Fall von Valsartan wurden z. B. noch Alkylamide gefunden, die zwar nicht toxisch sind, aber nicht Bestandteil des Wirkstoffes sein sollten [35]. |
Literatur
[2] https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/ Guidelines/Quality/Q3C/Q3C-R7_Document_Guideline_2018_1015.pdf
[3] https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/ Quality/ Q3D/ Q3D_Step_4.pdf
[4] Buschmann H, Holzgrabe U. Dtsch Apoth Ztg 2018;158:2898-2902 und 2019;159:50-54
[5] Keshav D, Desai S, Rathod D, Parikh C, Mokal R. An improved process for preparing losartan potassium, WO2010029457A2
[7] Pandey B, Lohray VB, Lohray BB. Process to prepare pioglitazone via several novel intermediates, US8173816B2
[8] Halama A, Hejtmankova L, Lustig P, Richter J, Srsnova L, Jirman J. Method for obtaining pioglitazone as an antidiabetic agent, US20050043360A1
[9] Meguro K, Fujita T. Thiazolidinedione derivatives, useful as antidiabetic agents, U.S. Patent 1987;4:687,777
[11] Vohr HE, Toxikologie, Bd. 2: Toxikologie der Stoffe, Wiley-VCH 2010
[12] Konieczny M, Sosnovsky G. Novel Aspects in the Preparation of Phorone, Z. Naturforsch. 1978;33b:454-460.
[13] Quirk E, Doggett A, Bretnall A. Determination of residual acetone and acetone related impuritiesin drug product intermediates prepared as Spray DriedDispersions (SDD) using gas chromatography with headspaceautosampling (GCHS), J. Pharm. Biomed. Anal. 2014;96:37-44
[14] Stare M, Laniewski K, Westermark A, Sjögren M and Tian W. Investigation on the Formation and Hydrolysis of N,N-Dimethylcarbamoyl Chloride (DMCC) in Vilsmeier Reactions Using GC/MS as the Analytical Detection Method, Org. Process Res. Dev., 2009;13:857-862.
[15] Yang Q, Haney BP, Vaux A, Riley DA, Heidrich L, He P, Mason P, Tehim A, Fisher LE, Maag H, Anderson NG. Controlling the genotoxins ethyl chloride and methyl chloride formed during the preparation of amine hydrochloride salts from solutions of ethanol and methanol. Org. Process Res. Dev. 2009;13:786-791.
[16] Teasdale A, Delaney EJ, Eyley SC, Jacq K et al. A Detailed Study of Sulfonate Ester Formation and Solvolysis Reaction Rates and Application toward Establishing Sulfonate Ester Control in Pharmaceutical Manufacturing Processes, Org. Process Res. Dev. 2010;14:999-1007.
[17] Gerber C, Toelle H. What Happened: The Chemistry Side of the Incident with EMS Contamination in Viracept Tablets. Toxicol. Lett. 2009;190:248-253.
[18] Snodin DJ. Residues of genotoxic alkyl mesylates in mesylate salt drug substances: Real or imaginary problems? Regul. Toxicol. Pharmacol. 2006;45:79-90.
[19] Szekely G, de Sousa MCA, Gil M, Ferreira F, and Heggie W. Genotoxic Impurities in Pharmaceutical Manufacturing: Sources, Regulations, and Mitigation, Chem. Rev. 2015;115:8182-8229.
[20] European Medicines Agency. CHMP Assessment Report for Viracept; Doc.Ref.: EMEA/CHMP/492059/2007; European Medicines Agency: London, 2007.
[21] Pozniak A, Müller L, Salgo M, Jones JK, Larson P, Tweats P, Elevated D. Ethyl Methanesulfonate (EMS) in Nelfinavir Mesylate (Viracept, Roche): Overview. AIDS Res. Ther. 2009;6:18.
[22] Patterson DE, Powers JD, LeBlanc M, Sharkey T, Boehler TE, Irdam E, Osterhout MH. Development of a Practical Large-Scale Synthesis of Denagliptin Tosylate. Org. Process Res. Dev. 2009;13:900-906.
[23] Elder DP, Delaney E, Teasdale A, Eyley S, Reif VD, Jacq K, Facchine KL, Schulte ostrich R, Sandra P, and David F. The Utility of Sulfonate Salts in Drug Development, J. Pharm. Sci. 2010;99:2948-2961
[24] Europäisches Arzneibuch, 9. Auflage, Deutscher Apotheker Verlag, Govi-Verlag, Pharmazeutischer Verlag, 2017
[25] Reynolds JEF, Parfitt K, Martindale K. The Extra Pharmacopoeia, 30th ed.; Pharmaceutical Press: London, 1989; p 1406.
[26] Schülé A, Ates C, Palacio M, Stofferis J, Delatinne JP, Martin JPB, Lloyd S. Monitoring and Control of Genotoxic Impurity Acetamide in the Synthesis of Zaurategrast Sulfate. Org. Process Res. Dev. 2010;14:1008-1014.
[27] Brown AD, Davis RD, Fitzgerald RN, Glover BN, Harvey KA, Jones LA, Liu B, Patterson DE, Sharp MJ. Process Development for Sodelglitazar: A PPAR Panagonist. Org. Process Res. Dev. 2009;13:297-302.
[28] Johnson DS, Li JJ, The Art of Drug Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, 2007;p108.
[29] Jackson RJ, Cooper KL, Tappenden P, Rees A, Simpson EL, Read RC, Nicholson KG. Oseltamivir, Zanamivir and Amantadine in the Prevention of Influenza: A Systematic Review, J. Infect. 2011;62:14.
[30] Drugs.com., Armodafinil, http://www.drugs.com/monograph/armodafinil.html, accessed April 28, 2019.
[31] Kaspi J, Leman O, Lexner J, Menashe N, Naddaka V, Saeed S. Process for the Preparation of Diphenylmethylthioacetamide. European Patent EP1260501, November 27, 2002.
[32] Dondre VG, Ghugare PD, Karmuse P, Singh D, Jadhav A, Kumar A. Identification and characterization of process related impurities in chloroquine and hydroxychloroquine by LC/IT/MS. LC/TOF/MS and NMR. J. Pharm. Biomed. Anal. 2009;49:873-879.
[33] Constable DJC, Jimenez-Gonzalez C, Henderson RK. Perspective on Solvent Use in the Pharmaceutical Industry, Org. Process Res. Dev., 2007;11:133-137.
[34] Marı´n A, Espada A, Vidal P, Barbas C. Major Degradation Product Identified in Several Pharmaceutical Formulations against the Common Cold, Anal. Chem. 2005;77:471-477.
[35] Sörgel F, Kinzig M, Abdel-Tawab M, Bidmon C, Schreiber A, Ermel S, Wohlfart J, Besa A, Scherf-Clavel O, Holzgrabe U. The contamination of valsartan and other sartans. Part 2. Untargeted Screening reveals contamination with amides additionally to known nitrosamine impurities. J. Pharm. Biomed. Anal. 2019;172:278-284.
Autoren
Dr. Helmut Buschmann hat in Aachen Chemie studiert. Nach Stationen bei Grünenthal und Esteve ist er Leiter der Chemie und Pharmazeutischen Entwicklung bei AiCuris in Wuppertal und Mitinhaber der Research, Development & Consultancy GmbH in Wien.
Prof. Dr. Ulrike Holzgrabe hat Chemie und Pharmazie studiert und ist seit 1999 Lehrstuhlinhaberin für Pharmazeutische und Medizinische Chemie in Würzburg.
Dr. Dirk Jung hat in Kaiserslautern Chemie studiert, und nach verschiedenen Stationen in der Entwicklung ist er Leiter der Qualitätseinheit bei der Arevipharma GmbH.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.