














- DAZ.online
- DAZ / AZ
- DAZ 34/2018
- Hoffnungsträger
Seltene Erkrankungen
Hoffnungsträger Edaravone
Zulassungsantrag für neues ALS-Medikament bei der EMA eingereicht
Mit einer Inzidenzrate von zwei bis drei Neuerkrankungen pro 100.000 Menschen pro Jahr zählt die amyotrophe Lateralsklerose zu den seltenen Krankheiten. Dennoch versterben jedes Jahr weltweit etwa 30.000 Menschen an dieser bislang unheilbaren Erkrankung. In Deutschland leben ca. 8000 ALS-Patienten, jedes Jahr kommen etwa 2250 dazu. Männer sind eineinhalb bis zweimal so häufig betroffen wie Frauen. Der Ausbruch der Krankheit erfolgt meistens zwischen dem 40. und 70. Lebensjahr. Frühe Symptome wie Muskelschwäche oder leichte Ermüdbarkeit der Muskulatur werden häufig nicht als ALS-Symptome erkannt. Deshalb vermutet man einen viel früheren Krankheitsbeginn.
Ice Bucket Challenge
Foto: imago/ZUMA Press
Die „ALS Ice Bucket Challenge“ (Eiswasserkübel-Herausforderung) war eine Spendenaktion zugunsten von Patienten mit amyotropher Lateralsklerose, die Mitte 2014 in den USA initiiert wurde. Sie verbreitete sich rasch über die sozialen Medien und war nach wenigen Wochen auch in Europa angekommen. Wer sich dieser Herausforderung stellen wollte, musste sich einen mit eiskaltem Wasser und Eiswürfeln gefüllten Eimer über den Kopf gießen oder gießen lassen, davor oder danach auf die Spendenaktion hinweisen sowie selbst etwas spenden. Anschließend nominierte er oder sie eine oder mehrere Personen, innerhalb von 24 Stunden die gleiche Prozedur durchzuführen. Die Idee hinter dem Eiswasserguss war, dass der Kandidat dadurch für einige Sekunden das Gefühl der Lähmung spüren sollte, unter dem ALS-Betroffene nach Ausbruch der Erkrankung mehrere Jahre leiden müssen. Häufig wurde der Kandidat gefilmt und das Video auf facebook oder YouTube veröffentlicht. Die Aktion bescherte vielen ALS-Kliniken, Verbänden und Patientenorganisationen weltweit umfangreiche finanzielle Mittel. Auch die ALS-Ambulanz an der Charité Berlin registrierte in dieser Zeit ein wesentlich höheres Spendenaufkommen als üblich: Innerhalb eines Jahres kamen dort ca. 1,6 Millionen Euro an Spendengeldern zusammen.
Progredienter Verlauf
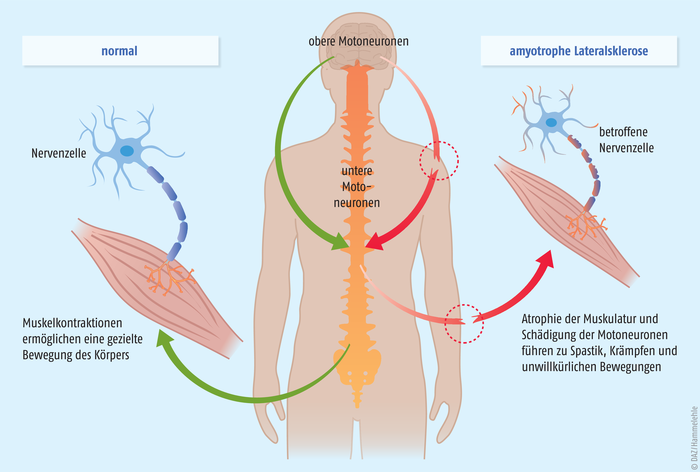
Die genaue Pathogenese der amyotrophen Lateralsklerose ist noch nicht geklärt. Zu den Ursachen existieren verschiedene Hypothesen, beispielsweise Autoimmunreaktionen, Virusinfektionen oder Umweltgifte. Daneben werden auch ein Überschuss des exzitatorischen Neurotransmitters Glutamat, oxidativer Stress, Entzündungsreaktionen oder eine abnorme Speicherung bestimmter Proteine in Gehirn und Rückenmark für die Pathogenese verantwortlich gemacht. Letztendlich kommt es durch die Degeneration der motorischen Nervenzellen (Motoneurone, s. Abb.) zum fortschreitenden Verlust der willkürlichen Muskelfunktion, zu Muskelzucken (Faszikulieren), Spastik und Lähmungen. Innere Organe sowie die kognitiven Fähigkeiten der Patienten sind nicht direkt betroffen. In den meisten Fällen tritt drei bis fünf Jahre nach der Diagnose der Tod durch Atemlähmung ein.
Wenige Behandlungsoptionen
Die Behandlung der ALS ist komplex, da die Methoden flexibel an die Bedürfnisse der Patienten angepasst werden müssen. Als beste Versorgungsform gilt die Betreuung durch eine ALS-Spezialambulanz, in der die verschiedenen Behandlungen wie Physiotherapie, Ergotherapie, Logopädie, Ernährungs- und Beatmungstherapie gut koordiniert werden können. Medikamentös kann die amyotrophe Lateralsklerose neuroprotektiv oder symptomatisch behandelt werden. Bisher war Riluzol (Rilutek® und Generika) das einzige Medikament, das das Fortschreiten der Erkrankung nachweislich zu verzögern vermochte. Riluzol erhöhte in Studien dosisabhängig die Wahrscheinlichkeit, das erste Therapiejahr zu überleben, um rund sechs bis 12%. Der genaue Wirkmechanismus von Riluzol ist noch nicht geklärt; man vermutet eine Hemmwirkung auf die glutamaterge Erregungsleitung. Zur symptomatischen bzw. palliativen Behandlung der amyotrophen Lateralsklerose werden verschiedene Wirkstoffe eingesetzt (s. Tab.).
Symptom/Anwendungsgebiet* |
Wirkstoffe |
|---|---|
respiratorische Insuffizienz |
chronische Phase: Mukolytika, Theophyllin (bei Obstruktion), nicht-kardioselektive Betablocker (z. B. Propranolol)Terminalphase: Morphin s.c. |
Dyspnoe (akut) |
Fentanyl (Nasenspray) |
Angstzustände/Panikattacken |
Lorazepam, Midazolam, Diazepam (auch als Suppositorien), SSRI (z. B. Paroxetin) |
Pneumonieprophylaxe |
N-Acetylcystein, Guaifenesin, Betablocker (Metoprolol oder Propranolol), Anticholinergika (Ipratropium) oder Theophyllin |
Hypersalivation |
Scopolamin (TTS), Amitriptylin, Atropin (1% Tropfen), Botulinumtoxin A oder B |
Thromboseprophylaxe |
niedermolekulare Heparine |
Depression |
Amitriptylin, SSRI |
pseudobulbäre Affektstörung (pathologisches Lachen und Weinen) |
Amitriptylin, eventuell SSRI (z. B. Fluvoxamin)Dextromethorphan + Chinidin (als Nuedexta® in Europa seit 2013 zugelassen, in Deutschland noch nicht im Markt) |
Schmerzen |
initial NSAR, später Opioide nach WHO-Schema |
Muskelkrämpfe/Faszikulationen |
Magnesium, Chininsulfat, Carbamazepin |
Spastik |
Antispastika, bei der PLS** auch L-Dopa;Botulinumtoxin A, Tetrahydrocannabinol (THC) + Cannabidiol als Oromukosalspray |
* nicht extra gekennzeichnet: Einige Wirkstoffe werden bei der ALS off label eingesetzt. **PLS: primäre Lateralsklerose; nur das obere Motoneuron ist betroffen | |
Hoffnungsträger Edaravone
Riluzol war bereits 1996 zur ALS-Behandlung zugelassen worden. Nach rund 20 Jahren steht nun mit dem Wirkstoff Edaravone in einigen Ländern eine neue Option zur Verfügung. Seit 2015 ist er unter dem Handelsnamen Radicut® in Japan und Südkorea verfügbar. Im Mai 2017 wurde Edaravone als Radicava® in den USA zugelassen. Bei der europäischen Zulassungsbehörde EMA ist seit Juni diesen Jahres ein Zulassungsantrag für Edaravone in der Bearbeitung, der von der Firma Mitsubishi Tanabe Pharma Europe Ltd eingereicht wurde. Bereits 2015 hatte die EMA den Orphan-Drug-Status erteilt. Bis Edaravone in Deutschland verfügbar ist, kann Radicut® über internationale Apotheken aus Japan importiert werden. Die gesetzlichen und privaten Krankenversicherungen übernehmen auf Antrag die Kosten der Behandlung.
Nicht für alle ALS-Patienten geeignet
Die Zulassungen von Edaravone in Japan, Südkorea und den USA beruhen auf zwei klinischen Studien aus Japan. In der Studie MCI-186-16 war Edaravone in der Gesamtpopulation der eingeschlossenen ALS-Patienten nicht wirksam. In einer Subgruppenanalyse ergab sich, dass der Wirkstoff das Fortschreiten der Erkrankung nur bei Vorliegen bestimmter klinischer Merkmale verlangsamen kann. Dazu zählen unter anderem eine Erkrankungsdauer von weniger als zwei Jahren, eine gute Atemfunktion und eine geringe bis mittlere Krankheitsprogression. In einer zweiten klinischen Studie (MCI-186-19) waren diese Merkmale als Einschlusskriterien definiert worden. Innerhalb einer 24-wöchigen Behandlung zeigte sich eine statistisch signifikante Verlangsamung der Krankheitsprogression in der Edaravone-Gruppe (n = 68) im Vergleich zur Placebo-Gruppe (n = 66) um etwa 30%, ermittelt mithilfe der ALS Functional Rating Scale (ALS-FRSr). ALS-FRSr ist eine international etablierte Skala zur Erfassung der ALS-Erkrankungsschwere, in der zwölf motorische Funktionen (Subskalen) bewertet werden, die typischerweise bei der amyotrophen Lateralsklerose beeinträchtigt sind. Die häufigsten Nebenwirkungen von Edaravone waren Blutergüsse, Gangstörungen und Kopfschmerzen. Außerdem sind nach Beendigung der Infusion lebensbedrohliche allergische Reaktionen auf den Wirkstoff oder den Hilfsstoff Natriumbisulfit mit Symptomen wie Urtikaria, Schwellungen in den Atemwegen und Atemnot möglich.
Nutzen-Risiko-Abwägung
Edaravone wird einmal täglich als 60-minütige Infusion verabreicht. Die Behandlung von ALS-Patienten erfolgt in vierwöchigen Therapiezyklen. Im ersten Behandlungszyklus sind 14 Tage lang einmal täglich eine Infusion vorgesehen, gefolgt von 14 Tagen Therapiepause. Ab dem zweiten Behandlungszyklus, der ebenfalls 28 Tage umfasst, erhalten die Patienten nur in den ersten zehn Tagen eine Edaravone-Infusion. Bei der erstmaligen Applikation muss der Patient in einer tagesklinischen oder stationären Einrichtung kontinuierlich überwacht werden. Der sich daraus ergebende zeitliche und logistische Aufwand stellt für Patienten mit krankheitsbedingt geringen körperlichen und psychischen Ressourcen eine starke Belastung dar. Deshalb sind die Ärzte gehalten, Nutzen und Risiken einer Behandlung mit Edaravone sorgfältig gegeneinander abzuwägen.
Orale Edaravone-Darreichnungsform in der Entwicklung
Das private Biotec-Unternehmen Treeway in den Niederlanden entwickelt derzeit eine orale Darreichungsform von Edaravone. In einer randomisierten Phase-I-Crossover-Studie mit 18 gesunden Probanden zeigte sich, dass sich mit dem Prüfmedikament TW001 mit einer Dosis von 140 mg eine größere Bioverfügbarkeit erzielen ließ als mit einer einstündigen Infusion von 60 mg Radicava®. Treeway plant derzeit eine Phase-III-Studie mit oralem Edaravone.
Ein weiterer potenzieller Wirkstoff zur ALS-Behandlung ist der Protein-Kinase-Inhibitor (PKI) Masitinib. Er ist bereits als Tierarzneimittel zur Therapie von Mastzelltumoren bei Hunden zugelassen. Als Wirkmechanismus postuliert man eine Hemmung von Mikrogliazellen, die bei einer amyotrophen Lateralsklerose wahrscheinlich eine pathologische Bedeutung haben. In einer 48-wöchigen Phase-II/III-Studie wurde Masitinib in Kombination mit Riluzol gegen Placebo plus Riluzol erfolgreich getestet. Diese Studie ist bereits abgeschlossen, weitere Untersuchungen sollen folgen. |
Literatur
Amyotrophe Lateralsklerose (Motoneuronerkrankungen). Leitlinie der Deutschen Gesellschaft für Neurologie (DGN), vollständig überarbeitet, Stand 1. Juni 2014, gültig bis 31. Mai 2019
Fachinformation Rilutek® 50 mg Filmtabletten, Stand Dezember 2013
Informationen der Deutschen Gesellschaft für Muskelkranke e.V. (DGM), www.als-selbsthilfe.de, Abruf am 24. Juli 2018
Doble A. The pharmacology and mechanism of action of riluzole. Neurology 1996;47:233-241
Bryson HM et al. Riluzole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in ALS. Drugs 1996;52(4):549-563
Applications for new human medicines under evaluation by the COMP. EMA/382940/2018 - Corr, www.ema. europa.org, Abruf am 24. Juli 2018
Edaravone bei der ALS – Update. Handlungsempfehlung für die Therapie mit Edaravone. www.als-charite.de/edaravone-bei-der-als-update/, Abruf am 24. Juli 2018
Treeway Announces Promising Data from Phase I Clinical Trial of Lead Program TW001 for ALS. Pressemitteilung von Treeway vom 29. Mai 2018, www.treeway.nl
Fachinformation Radicava® for intravenous use. Mitsubishi Tanabe Pharma Corporation, Stand August 2017
Petrov D et al. ALS Clinical Trials Review: 20 years of failure. Are we any closer to registering a new treatment? Front Aging Neurosci 2017;9:68, doi: 10.3389/fnagi.2017.00068
Public summary of opinion on orphan designation. Edaravone for the treatment of amyotrophic lateral sclerosis, Pressemitteilung des COMP vom 23 Februar 2015, EMA/COMP/744271/2014, www.ema.europa.eu, Abruf am 23. Juli 2018
AB Science announces positive top-line results of final analysis from study AB10015 of masitinib in amyotrophic lateral sclerosis (ALS). Pressemitteilung der AB Science vom 20. März 2017, www.ab-science.com Abruf am 23. Juli 2018



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.