

















- DAZ.online
- DAZ / AZ
- DAZ 34/2018
- Hilfe für die wenigen

Foto: Ralf Geithe – stock.adobe.com
Seltene Erkrankungen
Hilfe für die wenigen
Noch immer fehlen Orphan Drugs
Um die unterschiedlichen nationalen und internationalen Lösungsansätze für neue Therapieansätze für seltene Erkrankungen zu analysieren, bedarf es einer Definition eines Orphan Drugs. Diese erfolgt dabei über das jeweilige Indikationsgebiet. Orphan Drugs sind indiziert für seltene Erkrankungen bzw. Orphan Diseases. Die Namensgebung Orphan (englisch für Waise) unterstreicht, dass die Krankheiten lange Zeit nicht im Interesse von Wissenschaft und Industrie lagen; sie waren „verwaist“. Eine weltweit einheitliche Definition, wann eine Krankheit als selten gilt, gibt es dabei nicht. Während in der Europäischen Union (EU) ein Anteil an der Bevölkerung betrachtet wird, sind in den USA und Japan die absoluten Zahlen an Erkrankten relevant. Die Prävalenz, als selten zu gelten, liegt dabei zwischen 0,39 bis 0,63‰ (s. Tab. 1) [1, 2]. Die Zahl an Erkrankten ist gering, wenn man die Einzelkrankheiten betrachtet. Es gibt jedoch über 7000 bekannte Orphan Diseases. Die Zahl der insgesamt Betroffenen ist somit um ein Vielfaches größer und liegt in den USA und Europa bei ungefähr 30 Millionen Menschen [3, 4]. Das entspricht in der EU 6 bis 8% aller Bürger (s. Abb. 1). Dennoch ist das Wissen um diese Krankheiten wegen der individuellen Seltenheit in der Öffentlichkeit gering. Die meisten Erkrankungen sind in der Allgemeinheit völlig unbekannt und auch bei den Heilberufen häufig nicht im Bewusstsein. Einige wenige sind besser bekannt wie z. B. Mukoviszidose oder Leukämie; andere gelangen durch besondere Ereignisse oder Werbemaßnahmen in das öffentliche Interesse. Im Fall der amyotrophen Lateralsklerose (ALS), die durch den Tod von Stephen Hawking oder den Spendenaufruf unter dem Titel Icebucket-Challenge in den Blickpunkt von Medien rückte, gelang es so, erhöhte Aufmerksamkeit zu erlangen.
Region |
Definition |
Bevölkerungsanteil in ‰ |
|---|---|---|
Europäische Union |
< 5 von 10.000 EU-Bürgern |
< 0,5 |
USA |
< 200.000 US-Bürger |
< 0,64 |
Japan |
< 50.000 Japaner |
< 0,39 |
Russland |
< 19 von 100.000 Russen |
< 0,19 |
China |
< 76 von 100.000 Chinesen |
< 0,76 |
Aufmerksamkeit und damit das Wissen um solche Krankheiten spielen bei der Diagnose eine zentrale Rolle. Häufig dauert es Jahre, bis die richtige Diagnose gestellt wird. Dies liegt vor allem an der simplen Tatsache, dass Seltenes weniger wahrscheinlich ist und somit nicht mitbedacht und nicht erkannt wird. Ist dann eine Diagnose erstellt, steht der behandelnde Arzt häufig vor dem Problem, kein Arzneimittel mit dieser Indikation finden zu können. Nur für 5% der Orphan Diseases gibt es eine passende medikamentöse Therapieoption [3]. Ist ein Orphan Drug auf dem Markt, erhält es beschleunigten Marktzutritt für eine Behandlungsoption und wird zumeist rasch in die Leitlinien aufgenommen (s. Tab. 2).
Erkrankung |
Prävalenz in ‰ |
Medikation nach Leitlinie |
|---|---|---|
amyotrophe Lateralsklerose |
0,05 |
Basistherapie: Riluzol®* |
Narkolepsie |
0,2 |
Modafinil, Na-Oxybat*, Methylphenidat |
pulmonale Hypertonie |
0,005 |
Kalium-Kanalblocker, PDE-5-Hemmer, Riociguat*, Selexipag*, Prostanoide, Endothelin-Rezeptor-Antagonisten (Ambrisentan* u. a.) |
* Orphan Drug | ||
In den USA hat man bereits Anfang der 80er-Jahre die besondere Situation der Orphan Drugs erkannt und 1983 über die Gesetzgebung den sogenannten Orphan Drug Act erlassen. Das Gesetz soll die Erforschung und Bekämpfung seltener Erkrankungen fördern. Hier sind die Grundlagen zur Definition und dem Zulassungsverfahren als Orphan Drug festgelegt. So muss ein Orphan Drug ein Arzneimittel sein, das für eine seltene schwerwiegende Krankheit indiziert ist. Der Orphan-Drug-Status kann auch nur für einzelne Indikationen gelten, selbst wenn weitere Indikationen nicht in die Definition einer seltenen Erkrankung fallen. Daraus ergeben sich, in den USA, zwei Möglichkeiten für die Zulassung als Orphan Drug: Neuzulassung oder Indikationserweiterung. Bei der Neuzulassung wird das Arzneimittel als Orphan Drug zugelassen, wenn die Indikation eine Orphan Disease betrifft. Es handelt sich dabei um neue Arzneimittel mit neuem Arzneistoff oder bekanntem Arzneistoff in neuer oder veränderter Arzneiform. Im Fall der Indikationserweiterung erhält ein bereits zugelassenes Arzneimittel den Orphan-Drug-Status. Der neue Status gilt in diesem Fall jedoch nur für die Indikationen aus dem Gebiet der Orphan Diseases. Der Vorteil, den der Status mit sich bringt, umfasst Steuererleichterungen, eine Reduktion der Zulassungs- und Status-Erhaltungsgebühren der US-amerikanischen Gesundheitsbehörde (Food and Drug Administration, FDA), finanzielle Unterstützung von klinischen Studien sowie eine Marktexklusivität für die Dauer von sieben Jahren (s. Tab. 3). Diese Marktexklusivität verhindert, dass die FDA in dieser Zeit weitere Arzneimittel mit demselben Wirkstoff oder Therapieprinzip für dieselbe Orphan Disease zulassen kann [5]. Die klinischen Testungen werden mit bis zu 400.000 US-Dollar jährlich über bis zu vier Jahre (Phase II und III) subventioniert [6]. Mit dem im Dezember 2016 erlassenen 21st Century Cure Act (kurz: Cure Act) wurden der FDA weitere Möglichkeiten gegeben, besser auf die Bedürfnisse von Patienten, der Pharmaindustrie sowie des wissenschaftlichen Fortschritts einzugehen und flexibler auf neue Situationen reagieren zu können. Verstärkt kann auf den Bedarf der Patienten eingegangen werden. Klinische Studien können modernisiert werden, um sie für die geringe Patientenzahl praktikabler zu machen. Ebenso soll die Patientensicht in den Entscheidungsprozess bei der Zulassung von Arzneimitteln Einzug erhalten. Darüber hinaus sind im Cure Act zwei Entwicklungsprogramme beschrieben, die den Fortschritt in der Arzneistoffentwicklung forcieren sollen, dazu zählen das Breakthrough Devices Program und die Regenerative-Medicine-Advanced-Therapy (RMAT). Mit der Aufnahme in eines der Programme kann eine schnellere Marktzulassung erreicht werden. Dazu muss es sich bei den Arzneistoffen bzw. Zell- oder Gewebeprodukten (die in der RMAT eingesetzt werden) um Therapien handeln, die gegen schwere oder lebensbedrohliche Krankheiten gerichtet sind oder die eine Lücke in der aktuellen Therapie schließen können. Das Zulassungsverfahren läuft in enger Abstimmung mit der FDA ab, sodass zusammen festgelegt werden kann, was und wie genau geprüft wird; es muss nicht ausschließlich auf randomisierte kontrollierte Studien (randomized controlled trial, RCT) zurückgegriffen werden [7 – 10]. In-vivo-Tiermodelle, In-vitro-Daten, Fallberichte und computergestützte Rechenmodelle können genutzt werden, um die benötigten Daten zu erzeugen [8, 10]. Durch die Möglichkeit, die Gesamtheit aller wissenschaftlichen Verfahren und Quellen, nicht nur durch randomisierte kontrollierte Studien, zur Evidenzfindung nutzen zu können, wird es leichter, Wirksamkeit, Unbedenklichkeit und Qualität nachzuweisen und damit das Zulassungsverfahren zu beschleunigen; Kosten werden reduziert. Beispielsweise konnte im Fall von Ivacaftor, einem Arzneistoff zur Behandlung der Mukoviszidose, bei einer speziellen Mutation des Cystic Fibrosis Transmembrane Conductance Regulators (CFTR) die Indikation durch In-vitro-Daten auf weitere 23 Mutationen ausgeweitet werden [9].
Kriterium |
FDA |
EMA |
|---|---|---|
Marktexklusivität |
sieben Jahre |
zehn Jahre |
Nachprüfung |
keine |
nach fünf Jahren |
finanzielle Entlastung |
teilweise/ganz |
teilweise/ganz |
Überlegenheit gegen Standard |
nicht nachzuweisen |
nachzuweisen |
Indikationserweiterung |
möglich |
nicht möglich |
Zulassungsverfahren |
vereinfacht |
regulär |
In der EU wurde erst im April 2000 über die Europäische Arzneimittel-Agentur (EMA) eine entsprechende Verordnung zur Regelung von Orphan Drugs erlassen [11]. In der Verordnung wird zunächst beschrieben, was eine Orphan Disease ist und wie Orphan Drugs zugelassen werden. Dabei unterscheiden sich die Kriterien für die Zulassung (verglichen mit den USA) vor allem in einem Punkt: Um eine Zulassung als Orphan Drug zu erhalten, muss die Überlegenheit gegenüber der existierenden Behandlung nachgewiesen werden. Im Vergleich zu „Nicht-Orphan-Drugs“ unterscheidet sich die Zulassung nicht. Qualität, Wirksamkeit und Unbedenklichkeit müssen auf gleiche Weise nachgewiesen werden. Dabei darf im Einzelfall der Umfang der klinischen Testungen von den normalen Regularien abweichen, wenn aufgrund der Seltenheit der Erkrankung keine genügend große Testgruppe erstellt werden kann.
Die Vorteile eines Orphan-Drug-Status sind mit denen in den USA vergleichbar. Steuererleichterungen und der Erlass von Administrationsgebühren verringern die Kosten. Eine Marktexklusivität wird ebenfalls gewährt. Diese gilt für zehn Jahre mit der Einschränkung, dass keine Versorgungsengpässe entstehen und bei keinem anderen Arzneimittel eine Überlegenheit nachgewiesen werden kann. Die Exklusivität besteht gegenüber allen ähnlichen Arzneimitteln. Dabei gilt ein Arzneimittel als ähnlich, wenn drei Kriterien erfüllt werden: gleiche molekulare Strukturen, gleicher Wirkmechanismus und gleiche therapeutische Indikation. Besteht zwischen zwei Produkten in einem oder mehreren Punkten ein signifikanter Unterschied, werden diese nicht als ähnlich bewertet [12]. Der Antrag auf Anerkennung als Orphan Drug kann jederzeit während der Entwicklung beim Committee for Orphan Medicinal Products (COMP) gestellt werden. Vor Markteinführung prüft das COMP noch einmal, ob die Kriterien für ein Orphan Drug gegeben sind. Ab diesem Zeitpunkt gelten, bei positiver Bewertung, zehn Jahre Marktexklusivität. Nach fünf Jahren auf dem Markt kann auf Antrag eines Mitgliedstaates eine Überprüfung des Status erfolgen. Dabei wird geprüft, ob weiterhin weniger als fünf von 10.000 Menschen betroffen sind und kein anderes Arzneimittel als überlegen bewertet wurde. Sind die Kriterien nicht mehr erfüllt, verfällt der Orphan-Drug-Status. Eine Indikationserweiterung eines bereits zugelassenen Arzneimittels kann in der EU nicht zu einem Orphan-Drug-Status führen. Anders als in den USA ist es nicht möglich, für nur eine Indikation den Orphan Drug-Status zu erhalten, wenn andere Indikationen in den Bereich von häufigen Erkrankungen fallen. Soll ein bekanntes Arzneimittel für eine Orphan-Disease-Indikation den Orphan-Drug-Status erhalten, ist dafür als eigenes Arzneimittel – nicht Arzneistoff – eine eigene Dosierung, klinische Testung, Vermarktung und Zulassung nötig. Für eine Indikationserweiterung eines Orphan Drugs auf eine weitere Indikation aus dem Bereich der Orphan Diseases ist dies nicht nötig.
Was wiegt mehr: Nutzen oder Risiko?
Im Vergleich der beiden Zulassungsverfahren ist eine unterschiedliche Priorisierung von Nutzen und Risiko zu erkennen. Während die EMA bekannte und bewährte Methoden nutzt, um ein Höchstmaß an Therapiesicherheit zu gewährleisten, gesteht die FDA den Entwicklerfirmen mehr Freiheiten in den Zulassungsverfahren zu, die jedoch mit höheren Risiken für Patienten verbunden sein können. In-vitro-Studien und Metaanalysen können wichtige und richtige Daten zur Einschätzung liefern, erreichen aber nicht das Evidenzlevel von randomisierten kontrollierten Studien. Der von der EMA geforderte Nachweis der Überlegenheit gegenüber bisherigen Therapieprinzipien kann sich in vielen Fällen als schwierig erweisen, denn die geringe Anzahl an Patienten macht das Erbringen eines signifikanten Ergebnisses schwierig und meist aufwendig.
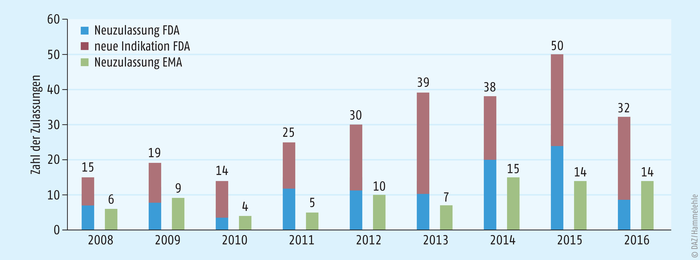
Der Vorsprung der USA von fast 20 Jahren im Bereich der Orphan Diseases aus dem Jahr 1983 zum Jahr 2000 und das liberalere Zulassungsverfahren macht sich in der Zulassungsstatistik deutlich (s. Abb. 2) [13, 14]. In den USA werden zwei- bis dreimal so viele Orphan Drugs zugelassen wie in der EU. Dabei ist zu beachten, dass über die Hälfte der Neuzulassungen Indikationserweiterungen sind, das heißt zugelassene Arzneimittel (für häufige Erkrankungen), die nun eine Indikation im Orphan-Disease-Bereich erhalten haben. Diese werden in der EU nicht als Orphan Drugs angesehen, da sie bereits für eine häufige Erkrankung zugelassen sind. Die Zahl an völlig neuen Arzneimitteln hat sich in den letzten Jahren aneinander angepasst.
Weltweit ist das Interesse an Orphan Drugs gestiegen. So nahm in den USA die Anzahl an Orphan Drugs 47% der Neuzulassungen ein, in der EU waren es 43% und in Japan 37% [15]. Aktuell sind in der EU 105 (Stand Juli 2018) [16] und in den USA 449 (Stand 2017) [17] Orphan Drugs zugelassen. Dabei fällt auf, dass es sich dabei vor allem um Arzneimittel zur Krebstherapie handelt (s. Abb. 3). 1900 Arzneistoffe in ihren Entwicklungsphasen haben in der EU den Orphan-Drug-Status zugesprochen bekommen. Eine Zusammenstellung der EMA zeigt, dass dabei vor allem Arzneistoffe gegen sehr seltene Erkrankungen den Orphan-Drug-Status erhielten [14]. 89% der Arzneistoffe sollen gegen Krankheiten eingesetzt werden, die maximal drei von 10.000 (0,03‰) Menschen betreffen [14]. Ist die Prävalenz jedoch noch kleiner, sinkt das Forschungsinteresse deutlich [18]. Die Art und Häufigkeit der Krankheit haben einen gleichbleibend großen Einfluss auf die Intensität der Forschung und damit auf die Anzahl an Orphan Drugs.
Dabei spielen neben Ätiologie und Prävalenz auch Faktoren wie politische Förderung, Patienten- und Interessensverbände sowie die öffentliche Bekanntheit eine große Rolle. In der Öffentlichkeit besser bekannte Erkrankungen wie Mukoviszidose oder ALS erfahren eine erhöhte wissenschaftliche Beachtung [18], die mit der Entwicklung von Orphan Drugs korreliert [15]. Social Media [19] sowie Patientenorganisationen können maßgeblich helfen, die Bekanntheit einer Krankheit zu steigern. Hiervon profitieren nicht nur Forscher, sondern auch Ärzte und besonders Patienten. Krankheiten werden schneller erkannt, und Patienten fühlen sich in ihrer Situation nicht allein gelassen. Die Icebucket-Challenge schaffte es, mehr als 220 Millionen US-Dollar an Spenden zu generieren. Diese wurden bereits genutzt, um wichtige genetische Grundlagen entschlüsseln zu können. Wie Betroffene berichten, ist jedoch die Anteilnahme und das Interesse der Gesellschaft ein noch größerer Wert für sie [20]. Einige Beispiele zeigen, wie auch Patienten und ihre Familien aktiv die Forschung unterstützen (z. B. Chion Foundation bei Prader-Willi-Syndrom). Mit Crowdfunding-Projekten wird vermehrt eine Finanzierungsgrundlage für Forschung und Arzneimittelentwicklung gesammelt, wobei häufig der Wunsch besteht, nicht nur den eigenen Angehörigen zu helfen, sondern zukünftigen Erkrankten eine bessere Therapie zu ermöglichen [21].
In Deutschland hilft seit 2010 das „Nationale Aktionsbündnis für Menschen mit seltenen Erkrankungen“ (NAMSE) bei der Verbesserung von Diagnose und Therapie. In dem Aktionsbündnis sind über 25 Partner organisiert, darunter das Bundesgesundheitsministerium, der Verein „Allianz Chronischer Seltener Erkrankungen” (ACHSE e. V.) sowie verschiedene Spitzen- bzw. Dachverbände des Gesundheitswesens [22]. 2013 wurde ein Aktionsplan durch das NAMSE veröffentlicht. Darin enthalten sind unter anderem die Forderungen nach neuen Arzneimitteln, einer einheitlichen Kodierung der Erkrankungen und dem Einrichten von ambulanten Versorgungsnetzwerken. Zur besseren Information für Betroffene, Angehörige und Fachpersonal wurde das „Zentrale Informationsportal Seltene Erkrankungen“ ins Leben gerufen [23].
Fazit
Generell lässt sich eine positive Entwicklung im Bereich der Orphan Drugs und der Orphan Diseases erkennen. Das allgemeine Forschungsinteresse steigt. Es werden immer mehr Orphan Drugs zugelassen. Das politische und gesellschaftliche Engagement scheint sich auszuzahlen. Betrachtet man den Markt jedoch differenzierter, wird schnell deutlich, dass noch nicht alle Erkrankte von der Situation profitieren können. Für zahlreiche Krankheiten fehlen weiterhin die passenden Arzneimittel. Eine sich immer weiter entwickelnde Förderung, nicht nur aus dem politischen Bereich heraus, bleibt somit unerlässlich. Vergleicht man die Zulassungen bei den Orphan Drugs mit der Zahl an regulären Zulassungen, fällt eine hohe „Durchfallquote“ auf. Hier können neue Zulassungsansätze eine Option sein, die Zulassungsrate weiter zu erhöhen. Dabei zeigt die USA eine Vorreiterrolle. Wie sich die liberaleren Zulassungsregularien auf die Zahl an neuen Arzneimitteln und auch auf die Arzneimittelsicherheit auswirken, bleibt abzuwarten. Die Möglichkeit, flexibler auf den wissenschaftlichen Fortschritt eingehen zu können, wird für die Zukunft sicher vorteilhaft sein. Letztendlich darf bei allem marktwirtschaftlichen und wissenschaftlichen Innovationsdrang die Sicherheit der Patienten jedoch nicht gefährdet werden. Arzneistoffe mit hohem Gefährdungspotenzial sollten also nicht kritiklos abgesegnet werden, nur um eine Therapielücke schließen zu können. Nutzen-Risiko-Bewertungen bleiben die maßgeblichen Kriterien. |
Literatur
[1] Franco P. Orphan drugs: the regulatory environment. Drug Discov Today 2013;18:163-172
[2] Richter T et al. Rare Disease Terminology and Definitions—A Systematic Global Review: Report of the ISPOR Rare Disease Special Interest Group 2015, doi:10.1016/j.jval.2015.05.008
[3] Orphan Drugs in the United States. Quintiles IMS Institute 2017, https://rarediseases.org/wp-content/uploads/2017/10/Orphan-Drugs-in-the-United-States-Report-Web.pdf, Abruf: 2. Mai 2018
[4] Orphan drugs and rare diseases at a glance. European Medicines Agency (EMA) EMEA/290072/2007, 3. Juli 2007
[5] Patents and Exclusivity. Food and Drug Administration (FDA), www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/UCM447307.pdf, Abruf: 13. April 2018
[6] Office of the Commissioner C for D, E und R. Developing Orphan Products: FDA and Rare Disease Day, 28. Februar 2011, Food and Drug Administration (FDA), www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/ucm239698.htm, Abruf: 13. April 2018
[7] Commissioner O. Of the 21st Century Cures Act. Food and Drug Administration (FDA), https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/21stCenturyCuresAct/default.htm, Abruf: 13. April 2018
[8] Gottlieb S. How FDA Plans to Help Consumers Capitalize on Advances in Science. FDA Voice. Food and Drug Administration (FDA), https://blogs.fda.gov/fdavoice/index.php/2017/07/how-fda-plans-to-help-consumers-capitalize-on-advances-in-science/, Abruf: 13. April 2018
[9] Hunter NL, Rao GR, Sherman RE. Flexibility in the FDA approach to orphan drug development. Nat Rev Drug Discov 2017;16:737-738
[10] Development OP. Recommended Tips for Creating an Orphan Drug Designation Application. Food and Drug Administration (FDA) 2018, www.fda.gov/downloads/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation/UCM598981.pdf, Abruf: 14. März 2018
[11] Commission Regulation(EC) No 847/2000. Off J Eur Communities 2000:23
[12] European Medicines Agency - Orphan medicines - Marketing authorisation and market exclusivity. www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000392.jsp&mid=WC0b01ac058061f019, Abruf: 13. April 2018
[13] Lanthier M. Insights into Rare Disease Drug Approval: Trends and Recent Developments NORD Rare Diseases & Orphan Products Breakthrough Summit. Food and Drug Administration (FDA) 2017, www.fda.gov/downloads/forindustry/developingproductsforrarediseasesconditions/ucm581335.pdf, Abruf: 13. April 2018
[14] Orphan Medicines Figures 2000-2017. European Medicines Agency 2017, www.ema.europa.eu/docs/en_GB/document_library/Other/2015/04/WC500185766.pdf, Abruf: 13. April 2018
[15] Murakami M, Narukawa M. Matched analysis on orphan drug designations and approvals: Cross regional analysis in the United States, the European Union, and Japan. Drug Discov Today 2016;21:544-549
[16] Zugelassene Orphan Drugs. Verband Forschender Arzneimittelhersteller e. V. (vfa), www.vfa.de/de/arzneimittel-forschung/datenbanken-zu-arzneimitteln/orphan-drugs-list, Abruf: 13. April 2018
[17] Quintiles IMS Institute. Orphan Drugs in the United States - Providing context for use and cost. 2017
[18] Mizoguchi H, Yamanaka T, Kano S. Research and drug development activities in rare diseases: differences between Japan and Europe regarding influence of prevalence. Drug Discov Today 2016;21:1681-1689
[19] Milne CP, Ni W. The Use of Social Media in Orphan Drug Development. Clin The 2017;39:2173-2180
[20] Sohn E. Fundraising: The Ice Bucket Challenge delivers. Nature 2017;550:113-114
[21] The Scientist Staff. Families of Children with Rare Diseases Fuel Gene Therapy Research. The Scientist Magazine®. The Scientist 2018:38-45
[22] Nationales Aktionsbündnis – NAMSE – Nationales Aktionsbündnis für Menschen mit Seltenen Erkrankungen. www.namse.de/aktionsbuendnis.html, Abruf: 13. April 2018
[23] Orphan Disease: So wirkt der nationale Plan für seltene Erkrankungen. www.aerztezeitung.de/politik_gesellschaft/versorgungsforschung/article/958234/orphan-disease-wirkt-nationale-plan-seltene-erkrankungen.html, Abruf: 13. April 2018
[24] Ludolph AC. Leitlinien für Diagnostik und Therapie in der Neurologie Amyotrophe Lateralsklerose (Motoneuron-Erkrankungen). in Leitlinien für Diagnostik und Therapie in der Neurologie, Georg Thieme Verlag KG, 2012
[25] Olschewski HO et al. Diagnosis and Therapy of Chronic Pulmonary Hypertension. Pneumologie 2006;60,749-771
[26] Gerloff C. Leitlinie für Diagnostik und Therapie in der Neurologie Narkolepsie. in Leitlinien für Diagnostik und Therapie in der Neurologie, Georg Thieme Verlag KG, 2012
Autoren
Markus Falkenstein, Apotheker; Studium der Pharmazie an der Heinrich-Heine-Universität Düsseldorf; Praktisches Jahr an der Heinrich-Heine-Universität Düsseldorf im Arbeitskreis von Prof. Stark und in der Apotheke am Schauspielhaus Bochum; Approbation 2017; seit 2017 Doktorand am Institut für Pharmazeutische und Medizinische Chemie der Heinrich-Heine-Universität Düsseldorf im Arbeitskreis von Prof. Dr. Dr. h.c. Holger Stark
Holger Stark, Apotheker; Studium der Pharmazie an der Freien Universität Berlin, seit 2013 an der Heinrich-Heine-Universität Düsseldorf (W3) und seit 2015 Geschäftsführender Leiter der Pharmazie; Forschungsschwerpunkte: Neurotransmitter (Histamin, Dopamin und Glutamat) sowie Lipid-Signalling in Arachidon- und Sphingolipid-Kaskaden, bildgebende oder multitargeting Liganden mit Arzneistoffcharakter; Miterfinder des ersten selektiven Histamin-H3-Rezeptor-Antagonisten Pitolisant (Wakix®) gegen Narkolepsie als Orphan Disease; seit 2004 Herausgeber des Archiv der Pharmazie – Chemistry in Life Sciences



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.