















- DAZ.online
- DAZ / AZ
- DAZ 11/2018
- Nicht nur heiß, rot und ...

Foto: sakkmesterke – stock.adobe.com
Pathophysiologie
Nicht nur heiß, rot und geschwollen
Entzündungen im Visier
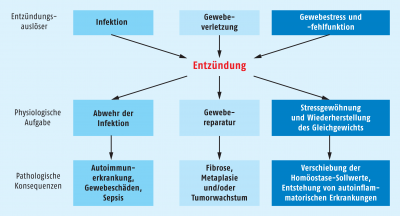
Kaum bemerkt und doch schnell passiert: Ein „Schnitt“ mit der Kante eines Papiers oder ein kleiner Holzsplitter, der sich in die Haut bohrt. Nach einem ersten kurzen Schmerz vergessen wir das kleine Malheur schnell wieder, bis die Stelle dann rot und dicker wird und der Körper mit einer Entzündung reagiert. Interessanterweise läuft ein ganz ähnlicher Prozess ab, wenn wir uns nach einer Verletzung eine Infektion einfangen oder wenn unser Körper mit bestimmten, sehr spezifischen Fehlfunktionen konfrontiert wird. Das Endziel dieser Reaktionen ist immer die Wiederherstellung des gesunden Gesamtstatus. Der Weg dahin geht über die Beseitigung von Fremdkörpern und Pathogenen und der Regeneration des geschädigten Gewebes – Prozesse, die jeden Tag ganz selbstverständlich und oft genug gänzlich unbemerkt ablaufen (Abb. 1). Wichtig dabei ist, dass die Entzündungsreaktion genauso geregelt wieder beendet wird, wie sie beim Auftreten einer akuten Gefahrensituation begonnen hat. Ist das nicht der Fall, richten sich die Prozesse mehr und mehr gegen gesundes Gewebe, das dann massiv in Mitleidenschaft gezogen wird.
Alle Grafiken: Zündorf
Wie funktioniert das alles in unserem Körper?
Generell lässt sich zusammenfassen, dass das Entzündungsgeschehen vermittelt wird von Induktoren, die von Sensoren wahrgenommen werden, was wiederum verschiedene Mediatoren auf den Plan ruft, die anschließend eine Vielzahl Effektoren ins Spiel bringen. Schlussendlich muss die ganze Reaktion – normalerweise – von speziellen Auflösungsfaktoren wieder beendet werden. Funktioniert dieser letzte Schritt nicht richtig und sind zu viele Mediatoren und Effektoren am Werk, kommt es zu länger anhaltenden Entzündungsreaktionen. Derartige Entgleisungen werden beispielsweise mit Alzheimer und anderen neurodegenerativen Erkrankungen in Verbindung gebracht, aber auch mit Tumoren oder verschiedenen Stoffwechselerkrankungen. Eine frühe antiinflammatorische Therapie könnte also extrem interessant sein, um bei diesen Krankheitsbildern rechtzeitig einzugreifen. Ansatzpunkte, eine Entzündungsreaktion zu blockieren, gibt es bereits reichlich, und es werden auch immer noch weitere entdeckt. Vor allem werden derzeit Mediatoren und Effektoren adressiert. Spannend in diesem Zusammenhang sind zudem die Auflösungsfaktoren.
Die Auslöser und ihre Sensoren
Exogene Faktoren
Mit dem stetig wachsenden Wissen um die zellulären Abläufe bei einer Entzündung zeigten sich sehr schnell die Ähnlichkeiten der Entzündungsprozesse mit den Aktivitäten des angeborenen Immunsystems.
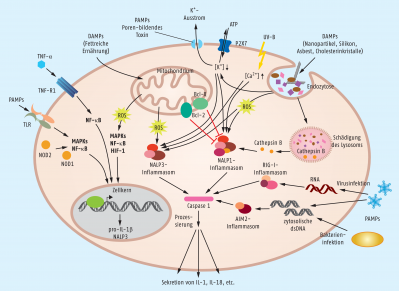
Bei einer Infektion mit einem Pathogen bringt ein Bakterium, ein Virus oder ein Pilz immer auch spezielle Strukturen mit, die von unserem Immunsystem als sogenannte pathogen-associated molecular patterns (PAMPs, Pathogen-assoziierte molekulare Muster) mithilfe der pattern-recognition receptors (PRR) erkannt werden. Ganz prominent sind in unserem Körper die membranständigen Toll-like-Rezeptoren (TLR) in diesen Prozess involviert, aber auch andere Rezeptoren wie z. B. die zytosolischen Rezeptoren NOD1 und NOD2 oder die Dectine 1 und 2 und etliche andere gehören zu diesen PRR. Das Akronym NOD leitet sich im Übrigen von nucleotide-binding oligomerization domain-containing protein ab. Diese Sensoren auf und in Zielzellen aktivieren nach Bindung der PAMPs intrazelluläre Signalkaskaden, was letztlich zur Bildung proinflammatorischer Effektoren führt (Abb. 2).
Ein zentrales intrazelluläres Signalmolekül ist hier der nukleäre Faktor κB (NF-κB), der nach seiner Aktivierung für die Expression proinflammatorischer Zytokine sorgt. Auch über die löslichen NOD-ähnlichen Rezeptoren (NOD-like receptors, NLR) im Cytoplasma wird NF-κB aktiviert. Zudem sind die Komplexe aus löslichem Rezeptor mit dem entsprechenden Liganden die Plattform für einen wichtigen, weiteren Protein-Komplex, der treffenderweise als Inflammasom bezeichnet wird. Dieser Protein-Komplex aktiviert Caspase-1, die daraufhin für die Bereitstellung des proinflammatorischen Zytokins IL-1β sorgt. Vermutet wird, dass Inflammasomen auch bei degenerativen Erkrankungen wie z. B. Alzheimer, Parkinson oder Atherosklerose vermehrt aktiviert werden, weshalb sie eine interessante Zielstruktur für die Therapie sind. Durch die verschiedenen löslichen Rezeptor-/Ligand-Komplexe, die am Aufbau des Inflammasoms mitwirken, ist eine ausgewogene Wirksamkeit bzw. Spezifität jedoch nicht ganz leicht zu erzielen.
Gerade Bakterien bringen oft eine stattliche Anzahl an Virulenzfaktoren mit in den Wirt ein, die üblicherweise nicht auf direkt passende Sensoren treffen. Vielmehr lösen sie meist indirekt eine Entzündungsreaktion aus, indem sie das Gewebe des Wirts schädigen, wodurch wiederum körpereigene Signale mobilisiert werden. Beispielsweise führen verschiedene Exotoxine von grampositiven Bakterien dazu, dass eine Pore in der Membran der Zielzelle gebildet wird. Dadurch kommt es zu einem ungewöhnlichen Ausstrom von K+-Ionen, der wiederum das sogenannte NALP3-Inflammasom (NACHT, LRR and PYD domains-containing protein 3-Inflammosome) aktiviert (Abb. 2).
Neben den mikrobiellen können noch andere exogene Faktoren eine Entzündung auslösen, wie z. B. der eingangs erwähnte Holzsplitter oder andere Fremdkörper, wobei hier vor allem Silikon- und Asbest-Partikel traurige Berühmtheit erlangt haben. Aber auch einige Allergene, Reiz- und Giftstoffe sind als auslösende Faktoren bekannt. Relativ große, nicht abbaubare Teilchen, wie sie in Form von Silikon- oder Asbest-Partikeln in den Körper gelangen, werden zwar von Makrophagen attackiert, allerdings gelingt eine Phagozytose meist schon allein wegen der Größe nicht. Stattdessen bilden sich größere Granulome aus Makrophagen und Fibroblasten, die permanent Entzündungssignale ausschütten. Die phagozytierten, unverdaubaren Partikel können jedoch auch zu einer Schädigung des Lysosoms führen, wodurch u. a. Cathepsin B freigesetzt wird, das wiederum Inflammasomen aktiviert (Abb. 2).
Endogene Faktoren
Endogene Entzündungsauslöser werden in Analogie zu den PAMPs auch als danger-associated molecular patterns (DAMPs) bezeichnet und entstehen z. B. nach einer akuten Verletzung, bei der Zellen beschädigt und z. B. ATP, K+-Ionen und verschiedene andere Moleküle aus dem Cytoplasma freigesetzt werden. ATP bindet daraufhin an Purin-Rezeptoren auf Makrophagen und trägt zur Aktivierung des Inflammasoms bei.
Andere Faktoren entstehen infolge von Stoffwechselstörungen aus unphysiologisch hohen Konzentrationen bestimmter Produkte wie z. B. Harnsäure oder Calciumpyrophosphat und führen letztlich zu Kristallablagerungen, die von Makrophagen ähnlich wie Fremdkörper bekämpft werden und chronische Entzündungen in Form von Gicht oder Pseudogicht auslösen. Zu dieser Art Entzündungs-Induktoren gehören auch oxidierte Lipoproteine aus Entgleisungen des Fettstoffwechsels oder infolge einer Hyperglykämie verstärkt auftretende, glykierte Proteine. Dazu kommen noch verschiedene endogene Liganden für Toll-like-Rezeptoren, die erst durch die Zerstörung biologischer Gewebeabgrenzungen oder der extrazellulären Matrix die entsprechenden Signalkaskaden induzieren können. Ein Beispiel sind hier Hyaluronan-Fragmente, die an den Toll-like-Rezeptor 4 binden können.
Die Liste möglicher, endogener Faktoren ist lang und wächst immer noch weiter. In der Prävention von Entzündungserkrankungen ist natürlich gerade die Vermeidung der DAMPs von großer Bedeutung.
Mediatoren und Effektoren
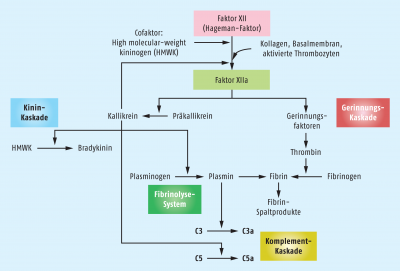
Eine wichtige Rolle im Entzündungsgeschehen spielt der Blutgerinnungsfaktor XII (Hageman-Faktor): Wurde initial Gewebe verletzt, kann der auf den Blutplättchen lokalisierte Faktor XII mit Kollagen und anderen Bestandteilen der extrazellulären Matrix interagieren, wird dadurch aktiviert und induziert anschließend nicht nur die Blutgerinnungskaskade, sondern auch die Kallikrein-Kinin-Kaskade, die Fibrinolyse-Kaskade sowie die Komplement-Kaskade (Abb. 3). Dabei entstehen verschiedene vasoaktive Fibrin-Abbauprodukte, Fibrin-Peptide A und B, die Komplementspaltprodukte C3a, C4a und C5a sowie Bradykinin. Ganz nebenbei werden die Thrombozyten selbst ebenfalls aktiviert und produzieren Entzündungsmediatoren wie Thromboxane oder Serotonin. Insgesamt wird eine ganze Reihe von Molekülen gebildet, die sich über ihre biochemischen Eigenschaften in sieben verschiedene Gruppen einteilen lassen und als Mediatoren der Entzündungsreaktion klassifiziert werden können (Tab. 1):
- vasoaktive Amine,
- vasoaktive Peptide,
- Fragmente von Komplementfaktoren,
- Lipidmediatoren,
- Zytokine,
- Chemokine
- proteolytische Enzyme.
Durch ihre vasodilatatorische und hyperalgesierende Wirkung kommt es zu den typischen Symptomen der Entzündung: Rötung, Schwellung, Schmerz und Wärme. Unter dem Einfluss dieser Mediatoren sezernieren aktivierte Immunzellen, vor allem die Makrophagen, Mastzellen und neutrophilen Granulozyten, zur Aufrechterhaltung der Entzündungsreaktion eine Reihe von proinflammatorischen Zytokinen, allen voran den Tumornekrosefaktor α (TNF-α) sowie die Interleukine 1 und 6 (IL-1 und IL-6), und locken darüber weitere Immunzellen an den Ort des Geschehens.
Mediatorklasse |
proinflammatorisch |
antiinflammatorisch |
|---|---|---|
Amine |
Histamin, Bradykinin |
Adrenalin, Noradrenalin |
Lipidmediatoren |
PGE2, PGI2, LTB4, LTC4
|
PGJ2, PGA1/2, Lipoxine
|
Komplement |
C3a, C5a |
C1q-Rezeptor |
zyklische Nukleotide |
cGMP |
cAMP |
Adhäsionsmoleküle |
E-Selectin, P-Selectin, ICAM1, VCAM1 |
avβ3-Integrin, Thrombospondin-Rezeptor, Phosphatidylserin-Rezeptor |
Zytokine |
TNF, IL-1β, IL-6 |
TGF-β, IL-10 |
Chemokine |
IL-8 (CCL8), GRO/KC, MIP1α (CCL3), MCP1 (CCL2) |
– |
Steroidhormone |
– |
Glucocorticoide |
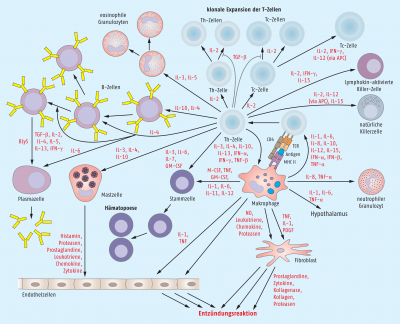
Die unterschiedlichen Zellen im Gewebe sind anschließend daran beteiligt, die normale Homöostase wiederherzustellen (Abb. 4).
Die Auflöser
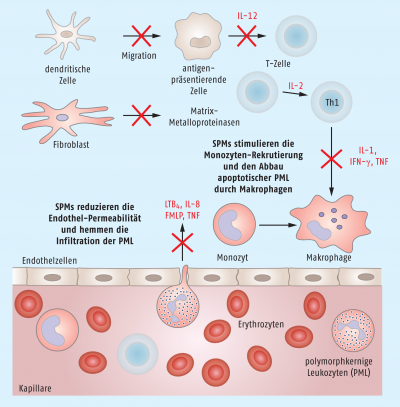
Hatte man lange Zeit angenommen, dass das Ende einer akuten Entzündung ein eher passiver Prozess ist, der durch das Fehlen der Induktoren und der allmählichen Elimination der Mediatoren und Effektoren abläuft, sind mittlerweile Faktoren bekannt, die sich aktiv an der Einstellung der normalen Gewebehomöostase und an der Beendigung der Entzündung beteiligen und zu den sogenannten specialized pro-resolving mediators (SPMs) gehören. Einige dieser Moleküle stammen von essenziellen, vielfach ungesättigten Fettsäuren (polyunsaturated fatty acids, PUFAs), omega-3-Eicosapentaensäure (EPA) und Docosahexaensäure (DHA) ab und werden als Lipoxine, Resolvine, Maresine und Protectine bezeichnet. Interessanterweise wird deren Bildung ebenfalls durch die proinflammatorisch wirkenden Prostaglandine (PG) E2 und D2 gesteuert. Neben ihrer Rolle als Auslöser von fiebrigen/schmerzenden oder allergischen Reaktionen induzieren die Prostaglandine E2 und D2 in Neutrophilen auch die Expression von 15-Lipoxygenase, die dann den Klassenwechsel in der Eicosanoid-Produktion weg von Leukotrienen und Prostaglandinen hin zu den Lipoxinen und Resolvinen in die Wege leitet. SPMs verringern die vaskuläre Permeabilität und verhindern dadurch die Einwanderung polymorphkerniger Nukleophiler in das entzündete Gewebe. Gleichzeitig stimulieren SPMs die Mobilisierung der Monozyten und sorgen dafür, dass Makrophagen die apoptotischen Neutrophilen wegräumen (Abb. 5).
Weitere Moleküle, die ebenfalls an der Auflösung der Entzündung beteiligt sind, sind z. B. Annexin A1, das adrenocorticotrope Hormon, Chemerin-Peptide und Galectin-1, aber auch Acetylcholin und andere Neuropeptide, Adenosin und gasförmige Mediatoren wie z. B. H2S und CO.
Therapeutika bei Entzündungsreaktionen
Entzündungen, die über den Holzsplitter oder die kleine Schnittwunde hinausgehen, sind unangenehm, aber doch üblicherweise selbstlimitierend und bedürfen keiner weiteren Behandlung mit Arzneimitteln. Sehr viel unangenehmer als diese kleinen sind jedoch größere akute oder sogar chronische Entzündungen, die uns z. B. in Form einer rheumatoiden Arthritis oder einer entzündlichen Darmerkrankung malträtieren.
Das Repertoire an Wirkstoffen, die in diese Prozesse eingreifen, ist inzwischen sehr umfangreich und zielt auf die unterschiedlichen Mediatoren und Effektoren des Reaktionsablaufs ab (Abb. 6, Tab. 2). Infolge des sehr einheitlichen Reaktionsablaufes, können somit mit einem Wirkstoff recht unterschiedliche Therapieziele erreicht werden: Quasi lassen sich mit einer Klappe gleich mehrere Fliegen schlagen. Die bisher eingesetzten Arzneistoffe zielen darauf ab, dass die Entzündungsmediatoren entweder nicht gebildet werden, indem ihre Expression bzw. Synthese inhibiert wird, oder aber die Entzündungsmediatoren ihre Wirkung nicht entfalten können, indem ihre Bindung an die Rezeptoren verhindert wird. Aber auch generellere Angriffspunkte werden adressiert, indem verhindert wird, dass sich die am Entzündungsgeschehen beteiligten Immunzellen vermehren können oder dass sie aus dem Blutkreislauf ins entzündete Gewebe auswandern.
Zielstrukturtyp |
spezifisches Target |
Wirkstoff (Beispiel) |
|---|---|---|
Enzyme |
COX 2 |
Coxibe |
COX 1 und COX 2 |
Diclofenac, NSAIDs |
|
PDE 4 |
Roflumilast, Apremilast |
|
JAK |
Baricitinib, Ruxolitinib, Tofacitinib |
|
IMPDH |
Mycophenolat-Mofetil |
|
G-Protein-gekoppelte Rezeptoren |
CysLT1 |
Montelukast, Zafirlukast |
S1P1 |
Fingolimod |
|
H1 |
Cetirizin, Desloratadin |
|
nukleäre Hormonrezeptoren |
Corticosteroide |
Glucocorticoide |
IL-17-Zytokine und Zytokin-Rezeptoren |
TNF-α und TNF-RII |
Infliximab, Adalimumab, Etanercept, Golimumab, Certolizumab |
IL-1β und IL-1RA |
Canakinumab, Anakinra |
|
IL-2 und IL-2R |
Basiliximab, Daclizumab |
|
IL-5 |
Mepolizumab, Reslizumab |
|
IL-6 und IL-6R |
Siltuximab, Sarilumab und Tocilizumab |
|
IL-12/23 |
Ustekinumab |
|
IL-17A und IL-17RA |
Secukinumab, Ixekizumab, Brodalumab |
|
IL-23 |
Guselkumab |
|
BLyS |
Belimumab |
|
Interferon α2 |
Peginterferon alfa-2a, Peginterferon alfa-2b |
|
Interferon β1 |
Interferon beta-1b |
|
Interferon γ |
Interferon gamma-1b |
|
Zellinteraktionsmoleküle (Zelladhäsionsmoleküle und kostimulatorische Moleküle) |
CD2 und LFA-3 |
Alefacept |
VLA-4 und CD49d |
Natalizumab |
|
α4β7-Integrin |
Vedolizumab |
|
CTLA-4-Ig |
Abatacept |
|
Oberflächenmoleküle zur B-/T-Zelldepletion über Induktion einer ADCC/CDC |
CD20 |
Rituximab, Ofatumumab, Obinutuzumab, Ibritumomab-Tiuxetan |
CD52 |
Alemtuzumab |
Hemmung der Mediator-Synthese
Natürlich muss in diesem Zusammenhang die Acetylsalicylsäure erwähnt werden, die bereits seit Ende des 19. Jahrhunderts als Hemmer der Cyclooxygenasen 1 (COX-1) und COX-2 und somit als Inhibitor der Prostaglandin-Synthese verfügbar ist. Seit der Zeit sind noch etliche weitere, ähnliche Wirkstoffe entwickelt worden, die zum Teil selektiv nur die COX-2 inhibieren. Mittlerweile weiß man, dass die Acetylsalicylsäure den Vorteil hat, dass sie ebenfalls die Bildung von Lipoxinen vermittelt und dadurch die natürliche Beendigung der Entzündungsreaktion unterstützt. Demgegenüber werden die Coxibe als „resolutionstoxisch“ bezeichnet, da sie zwar sehr effizient die Freisetzung der proinflammatorischen Prostaglandine verhindern, allerdings blockieren sie dadurch auch die Bildung der 15-Lipoxygenase. Dieses Enzym ist wiederum wichtig für den Wechsel hin zu den entzündungsauflösenden Lipoxinen.
Eine generelle Inhibition der Synthese von entzündungsrelevanten Enzymen sowie von proinflammatorischen Zytokinen und Chemokinen findet über die Glucocorticoide statt. Diese Wirkstoffe finden und binden ihre Rezeptoren zunächst im Zytosol der Zellen und wandern anschließend im Komplex in den Zellkern, wo sie die Expression proinflammatorischer Gene hemmen und die der antiinflammatorischen Gene fördern. Das Angebot an mittlerweile verfügbaren Glucocorticoid-Arzneistoffen deckt verschiedene systemische oder topische Anwendungsmöglichkeiten ab und bietet für jegliche Art von Entzündungsherd eine Therapieoption. Viel wird darüber spekuliert, wie sich letztlich die Wirksamkeit der alten Antimalariamittel Chloroquin und Hydroxychloroquin bei der Behandlung der rheumatoiden Arthritis erklären lässt. Da sich die beiden Substanzen in Lysosomen anreichern und dort den pH-Wert erhöhen, ist ein Erklärungsansatz die Störung der lysosomalen Enzyme und Inhibition des Toll-like-Rezeptors 9, der in der Membran der Endosomen/Lysosomen lokalisiert ist. Alternativ könnte über eine Fehlfunktion der Lysosomen auch das Recycling der Toll-like-Rezeptoren auf der Zelloberfläche, wie TLR1, TLR2 oder TLR4, beeinträchtigt sein. Das würde wiederum die nachgeschaltete Signalweiterleitung in die Zelle und die Synthese von Prostaglandinen und proinflammatorischen Zytokinen beeinträchtigen. Beobachtet wurde jedenfalls in Makrophagenzellkultur, dass die Zellen in Anwesenheit von Chloroquin weniger Entzündungsmediatoren produzieren.
Ob Sulfasalazin und Mesalazin ihre entzündungshemmende Wirkung eher über die Inhibition der Prostaglandin- und Leukotrien-Synthese oder aber über die Hemmung von NF-κB die Expression von z. B. TNF-α verhindern, ist nicht eindeutig geklärt. Sicher ist allerdings, dass sie wirken.
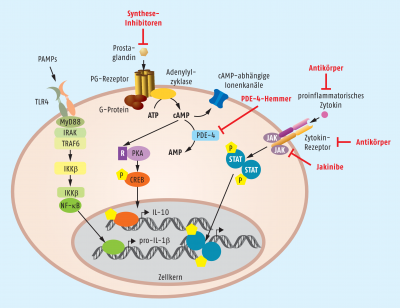
Ein Ansatz, der den Übergang zwischen der Hemmung der Mediator-Expression und der Hemmung einer Mediator-Wirkung aufzeigt, wird über die PDE-4-Inhibitoren Roflumilast und Apremilast verfolgt. Prostaglandine führen nach Kopplung an ihre Rezeptoren zu einer intrazellulären Signalweiterleitung, die zum Teil über die Bildung von zyklischem Adenosinmonophosphat (cAMP) verläuft. Dieser Botenstoff wiederum sorgt über verschiedene Wege für eine Expression antiinflammatorischer Zytokine und verhindert gleichzeitig die Bildung der proinflammatorischen Gegenspieler. In Zellen, die in einem entzündeten Gewebeverband liegen, wird Phosphodiesterase 4 exprimiert, die cAMP spaltet und so den proinflammatorischen Prozess fördert. Durch die gezielte Hemmung des Enzyms kann somit die Entzündung unterdrückt werden.
Direkte Hemmung der Mediator-Wirkung
Praktisch nicht mehr wegzudenken aus der Therapie chronisch entzündlicher Erkrankungen, wie beispielsweise rheumatoider Arthritis, Morbus Crohn oder Colitis ulcerosa, sind die Antagonisten des Tumornekrosefaktors α. Das Prinzip, den Mediator abzufangen und an der Bindung an seinen Rezeptor zu hindern, ist eigentlich logisch und einfach nachzuvollziehen. Zur Verfügung stehen bei TNF-α die Antikörper Infliximab, Adalimumab, Golimumab mit den zum Teil schon vorhandenen Biosimilars, das pegylierte Antikörper-Fragment Certolizumab Pegol sowie das Fusionsprotein aus Rezeptor und Antikörper-Fc-Teil Etanercept, für das ebenfalls bereits Biosimilars verfügbar sind. Nachdem die Blockade des Haupt-Entzündungsmediators TNF-α so erfolgreich war, wurde das prinzipielle Vorgehen auch für andere, sehr dominante proinflammatorische Zytokine kopiert. Die Wirkung des Interleukins 1 wird entweder durch den gegen IL-1β gerichteten Antikörper Canakinumab oder durch den IL-1-Rezeptorantagonist Anakinra aufgehoben. Mit Siltuximab ist ein Antikörper gegen IL-6 und mit Sarilumab und Tocilizumab sind zwei Antikörper gegen den IL-6-Rezeptor auf dem Markt, die spezifisch die Wirkung des Zytokins verhindern. Andere Antikörper-Moleküle, die nach dem gleichen Prinzip arbeiten, sind Ixekizumab und Secukinumab gegen IL-17A, Brodalumab gegen den IL-17-Rezeptor A, Guselkumab gegen IL-23 und Dupilumab gegen den IL-4-Rezeptor alpha, wobei die Indikationen der Wirkstoffe derzeit noch sehr unterschiedlich sind. Mit einer Annäherung der Indikationen kann allerdings relativ zeitnah gerechnet werden.
Mediatoren, an die man im Zusammenhang mit Entzündungen im Allgemeinen vielleicht nicht ganz so oft denkt, sind Histamin und Leukotrien, deren Bindung an ihre Rezeptoren ebenfalls sehr effizient verhindert werden kann. Die entsprechenden Wirkstoffe Cetirizin und Desloratadin bzw. Montelukast sind eher aus der Behandlung z. B. der allergischen Rhinitis vertraut.
Indirekte Hemmung der Mediator-Wirkung
Der große Nachteil der direkten Zytokin-Antagonisten ist ihr großes Molekulargewicht und ihre Protein-Natur, weshalb sie immer parenteral appliziert werden müssen. Einfacher wäre eine Therapie mit niedermolekularen, oral verfügbaren Inhibitoren. Nachdem viel über die Signalweiterleitung im Nachgang zur Zytokin-Rezeptor-Bindung bekannt war, wurden daraus einzelne Zwischenstufen als Zielstrukturen für antiinflammatorische Wirkstoffe untersucht. Relativ neue Arzneistoffe, die aus dieser Überlegung heraus entwickelt wurden, sind Inhibitoren der Janus-Kinasen (JAK). Einige der sogenannten Jakinibe, wie beispielsweise Baricitinib, Ruxolitinib und Tofacitinib sind bereits zugelassen. Die Janus-Kinasen sind Signalvermittler für viele Rezeptoren, die über keine eigene intrazelluläre Kinase-Aktivität verfügen. Dazu gehören beispielsweise die Rezeptoren für die Typ-I-Zytokine IL-2, IL-4, IL-7, IL-9, IL-15, und IL-21 sowie für die Typ-II-Zytokine wie z. B. α-, β- und γ-Interferone, IL-10, IL-19, IL-20, IL-22, IL-24 und IL-26. Insgesamt vier verschiedene Janus-Kinasen (JAK1, JAK2, JAK3 und TYK2 [Tyrosinkinase 2]) werden in den Zellen genutzt, um anschließend signal transducer and activator of transcription (STAT)-Proteine zu aktivieren, die dann wiederum die proinflammatorische Genexpression steuern. Die zugelassenen Jakinibe Baricitinib und Ruxolitinib sind relativ selektiv für JAK1 und JAK2, während Tofacitinib vor allem JAK1 und JAK3 inhibiert. Zu bedenken ist, dass Janus-Kinasen auch Signalwege anderer Rezeptoren steuern, worüber sich einige unerwünschte Arzneimittelwirkungen der Jakinibe erklären lassen.
Hemmung der Einwanderung der Effektorzellen in das entzündete Gewebe
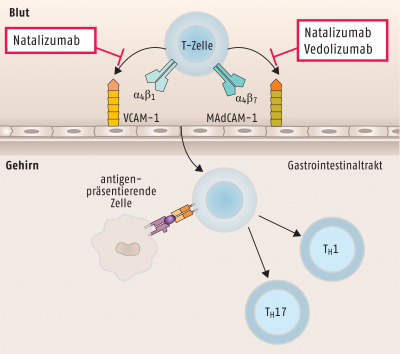
Zur Aufrechterhaltung der Entzündungsreaktion müssen Immunzellen aus dem Blut in das Gewebe auswandern. Dieser Vorgang wird über die Interaktion zwischen Oberflächenmolekülen auf den Immunzellen einerseits und den Gefäß-Endothelzellen andererseits initiiert. Wird diese Wechselwirkung durch sogenannte selektive Adhäsionsmolekül-Inhibitoren (selective adhesion molecule inhibitors) wie den Antikörpern Natalizumab oder Vedolizumab gestört, fehlt der Nachschub an Entzündungszellen im Gewebe. Während Natalizumab die α4-Untereinheit der verschiedenen Integrine auf den Immunzellen erkennt, bindet Vedolizumab sehr viel spezifischer an das α4β7-Integrin auf aktivierten T-Lymphozyten im Gastrointestinaltrakt, weshalb vermutet wird, dass unter Therapie mit Vedolizumab weniger Nebenwirkungen auftreten (s. Abb. 7).
Noch viel früher in der Verteilung der Immunzellen im Körper setzt Fingolimod an. Dieses Molekül ist ein funktioneller Sphingosin-1-Phosphat-Rezeptor-Antagonist, der die Lymphozyten im Lymphknoten hält und so die Anzahl der im Körper zirkulierenden Immunzellen reduziert, die dann auch nicht im entzündeten Gewebe zur Verfügung stehen.
Hemmung der Aktivierung und Proliferation von Immunzellen
Für Immunzellen gilt häufig das Prinzip, dass eine Aktivierung gleichzeitig ein Signal zur Proliferation ist. Für T-Lymphozyten läuft die Aktivierung über die Wechselwirkung zwischen T-Zellrezeptor und einem beladenen MHC-Komplex (major histocompatibility complex, Haupthistokompatibilitätskomplex) auf z. B. antigenpräsentierenden Zellen. Allerdings wird sicherheitshalber immer noch ein zweites Signal benötigt, das durch die Interaktion zwischen CD28/B7 auf den beiden Zelltypen zustande kommen muss. Das zytotoxische T-Lymphozyten-Antigen 4 (CTLA-4) verhindert dieses costimulatorische Signal und somit auch die T-Zell-Aktivierung. Mit Abatacept und Belatacept stehen zwei Fusionsproteine zur Verfügung, die jeweils aus der CTLA4-Bindungsdomäne mit einem Fc-Antikörperteil bestehen, und so in der Lage sind, die T-Zellaktivierung zu verhindern.
Das wichtigste Proliferationssignal für T-Lymphozyten ist Interleukin 2. Über verschiedene Mechanismen können die Immunsuppressiva Ciclosporin, Pimecrolimus, Tacrolimus, Everolimus und Rapamycin die Expression von IL-2 hemmen. Auf der anderen Seite findet über Daclizumab und Basiliximab eine Hemmung der IL-2-Wirkung auf T-Zellen statt. Die beiden Antikörper binden an eine Untereinheit des hochaffinen Rezeptors für Interleukin 2 auf aktivierten T-Zellen und inhibieren dadurch das Wachstumssignal.
Ein vergleichbarer Stimulus bei B-Zellen ist der B-lymphocyte stimulator (BLyS), der als „Überlebensfaktor“ für die Differenzierung in langlebige Plasmazellen verantwortlich ist. Mit Belimumab steht ein Antikörper zur Verfügung, der dieses Zytokin abfängt und dadurch letztlich die Lebensdauer der B-Lymphozyten verkürzt.
Der Erfolg der sogenannten disease modifying antirheumatic drugs (DMARDs) in der Behandlung chronischer Entzündungen wie der rheumatoiden Arthritis liegt darin begründet, dass einige von ihnen in die DNA-Synthese und somit in die Proliferation von Zellen eingreifen. Im Entzündungsprozess vermehren sich gerade aktivierte Immunzellen und werden dann besonders effizient von Methotrexat, Azathioprin oder Leflunomid getroffen.
Gezielte Zerstörung der Effektorzellen
Lymphozyten können nicht nur an ihrer Proliferation gehindert, sondern sogar relativ gezielt abgetötet werden. Vermittelt über Antikörper, die spezifisch an Moleküle auf der Oberfläche der T- und/oder B-Zellen binden, können das Komplementsystem und natürliche Killerzellen aktiviert werden, um die markierten Lymphozyten über Komplement-abhängige Zytotoxizität (complement-dependent cytotoxicity, CDC) bzw. Antikörper-abhängige zelluläre Zytotoxizität (antibody-dependent cellular cytotoxicity, ADCC) zu zerstören. Rituximab, Ofatumumab und Obinutuzumab sind alternative Moleküle, die selektiv an CD20 auf B-Zellen binden und genau diesen Effekt ausüben. Auch der radioaktiv markierte Antikörper Ibritumomab-Tiuxetan adressiert CD20, führt allerdings über die mitgebrachte Quelle für Beta-Strahlung zur Zerstörung der B-Zellen.
Sowohl B- als auch T-Zellen werden durch die Anwendung von Alemtuzumab eliminiert. Dieser Antikörper bindet an CD52 auf der Oberfläche der Lymphozyten und induziert sowohl die Komplement-abhängige Zytotoxizität als auch die Antikörper-abhängige zelluläre Zytotoxizität.
Ausblick
Angesichts der Vielzahl an therapeutischen Möglichkeiten, irgendwo innerhalb des Entzündungsgeschehens zu intervenieren, kann man kaum glauben, dass es noch neue Ansatzpunkte geben könnte.
Weit gefehlt: Gerade weil jetzt so viel über das Signalnetzwerk und die beteiligten Faktoren bekannt ist, kann man sich gezielt auf die Suche nach kleinen, eventuell sogar oral verfügbaren Wirkstoffen machen. In naher Verwandtschaft zu den Janus-Kinasen könnte vielleicht auch bald Fostamatinib als Inhibitor der spleen tyrosine kinase (Syk) zugelassen werden. In Japan und China gibt es Iguratimod als Arzneimittel bei Entzündungen. Dieses Molekül hemmt einerseits die Cyclooxygenase 2 und andererseits den wichtigen zentralen Transkriptionsfaktor NF-κB, der die Expression zahlreicher proinflammatorischer Zytokine steuert. Allerdings weiß man mittlerweile, dass NF-κB auch in der Resolution der Entzündungsreaktion eine wichtige Rolle spielt, was die Einsatzmöglichkeit eines entsprechenden Inhibitors vielleicht wieder etwas relativiert.
À propos Resolution: Hier gibt es noch richtig große weiße Flecken bezüglich Zielstrukturen für Entzündungshemmer. Mimetika von Molekülen, die an der Beendigung des Entzündungsprozesses beteiligt sind, wie Resolvin E1, Protectin PD1 und Maresin MaR1, befinden sich bereits in der klinischen Entwicklung. Und mit der Pyruvatkinase M2 oder dem IL-1-Rezeptor 8 könnten zwei ganz neue, interessante Targets gefunden worden sein, über deren Inhibition bzw. Aktivierung ebenfalls ein antiinflammatorischer Effekt erzielt werden kann.
Fazit
In der Gesamtschau ist die Anzahl der – im weitesten Sinne – antiinflammatorisch wirkenden Moleküle gewaltig. Allerdings muss man natürlich berücksichtigen, dass auch die Spanne, was alles noch zu „Entzündungsprozessen“ gezählt werden kann, extrem breit ist. Dadurch steht für nahezu jeden Schweregrad einer Erkrankung ein passender Wirkstoff zur Verfügung. Trotzdem werden viele Patienten noch nicht ausreichend versorgt, was sich vielleicht durch die Verfügbarkeit einiger Biosimilars ändern könnte. |
Literatur
Ghoreschi K, Gadina M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp Dermatol 2014;23:7-11
Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol 2002;2:787-795
Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 2007;7:31-40
Medzhitov R. Origin and physiological roles of inflammation. Nature 2008;454:428-435
Spadaccini M, D‘Alessio S, Peyrin-Biroulet L, Danese S. PDE4 Inhibition and Inflammatory Bowel Disease: A Novel Therapeutic Avenue. Int J Mol Sci 2017;18:pii:E1276, doi: 10.3390/ijms18061276
Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol 2016;7:160, doi: 10.3389/fimmu.2016.00160. eCollection 2016
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140:805-820
Autoren
Prof. Dr. Theo Dingermann ist Seniorprofessor am Institut für Pharmazeutische Biologie an der Goethe-Universität Frankfurt.
Dr. Ilse Zündorf ist dort als akademische Oberrätin tätig.
Institut für Pharmazeutische Biologie, Biozentrum, Max-von-Laue-Straße 9, 60438 Frankfurt/Main



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.