















- DAZ.online
- DAZ / AZ
- DAZ 6/2017
- Drei Eltern und ein Baby

Foto: Halfpoint - fotolia.com; Montage: DAZ/nd
Hintergrund
Drei Eltern und ein Baby
Eine außergewöhnliche Intervention zur Umgehung eines Gendefekts
Mitochondrien als „Troublemaker“
Wer interessiert sich schon für Mitochondrien? Natürlich weiß man, dass diese Organellen, die wahrscheinlich irgendwann in der Evolution von einer eukaryontischen Urzelle als bakterielle Überbleibsel eingefangen wurden, die Kraftwerke aller kernhaltigen Zellen bilden. Und man weiß auch, dass Mitochondrien ein eigenes Genom und eine eigene Proteinbiosynthese-Maschinerie besitzen. Weniger bekannt ist vielleicht, dass pro menschlicher Zelle 1000 bis 2000 dieser bizarren Partikel vorhanden sind, die allesamt und ausschließlich von der Mutter stammen.
Wenig überraschend ist es möglich, dass in der mitochondrialen DNA Mutationen vorkommen, die Funktionsstörungen der betroffenen Mitochondrien nach sich ziehen. Typischerweise treten solche Mutationen nicht in allen Mitochondrien auf. Fehlerhafte Mitochondrien geben ihre Mutationen jedoch an ihre „Nachkommen“ weiter, da sie sich innerhalb unserer Zellen durch Teilung vermehren. Betroffene Personen besitzen also in ihren Zellen in der Regel eine Mischung aus normalen und funktionsgestörten Mitochondrien, was als Heteroplasmie bezeichnet wird. Überschreitet der Prozentsatz an funktionsgestörten Mitochondrien eine bestimmte Schwelle, kann sich der Fehler in der mitochondrialen DNA phänotypisch bemerkbar machen und Ursache für teils schwere Erkrankungen sein. Dazu müssen in der Regel mehr als 60 bis 90% der Mitochondrien ein mutiertes Genom aufweisen [1]. In extremen Fällen sind sogar alle Mitochondrien eines Menschen von der Mutation betroffen; man spricht dann von Homoplasmie. Beim gesunden Menschen sind alle Mitochondrien identisch und nicht mutiert.
All das macht es plausibel, dass mitochondriale Krankheiten nicht nur extrem selten (1/5000 Geburten), sondern auch extrem heterogen sein müssen, sowohl hinsichtlich der Schwere der Krankheit als auch hinsichtlich der Organe, in denen die funktionellen Ausfälle erkennbar werden. Ebenfalls sehr heterogen ist der Beginn der Krankheit. Krankheitssymptome können bereits im Säuglingsalter offensichtlich werden, die Krankheit kann aber auch erst im Kleinkindalter oder gar im Erwachsenenalter ausbrechen. Krankheitsphänotypen sind retardiertes Wachstum, Taubheit, Erblindung, Funktionsstörungen am Herzen, der Leber und der Nieren, Lernschwäche, eine unterentwickelte Kontraktionsfähigkeit der Muskeln und teils schwere neurologische Probleme [2]. Für betroffene Frauen mit Kinderwunsch besteht dadurch das Problem, dass die genetische Präimplantationsdiagnostik an ihre Grenzen stößt: Es können keine sicheren Prognosen gestellt werden, ob und wie schwer das Kind erkranken wird.
Der konkrete Fall
Das jordanische Elternpaar, das sich zu dem experimentellen Eingriff entschieden hatte, hatte einen bemerkenswerten Leidensweg hinter sich. Die Mutter litt an einem seltenen genetischen Defekt der Mitochondrien, der Ursache für die Ausprägung eines Leigh-Syndroms war. Nach vier Fehlgeburten wurde 2005 ein Mädchen geboren, das jedoch im Alter von sechs Jahren an der Krankheit verstarb. Ein weiteres Kind lebte nur acht Monate.
Ein Leigh-Syndrom, das auch als subakute nekrotisierende Enzephalopathie bezeichnet wird, kann durch eine Vielzahl von Mutationen in unterschiedlichen mitochondrial, aber auch chromosomal kodierten Genen verursacht werden. Therapieoptionen stehen für mitochondrial verursachte Krankheiten nicht zur Verfügung. Daher versucht man, so gut wie möglich die Symptome zu behandeln [3].
In ihrer Not suchte das jordanische Ehepaar Rat und Hilfe bei einem amerikanischen Arzt, der dem Ehepaar einen experimentellen Lösungsansatz anbot, der nicht ganz korrekterweise als Mitochondrien-Spende (mitochondrial replacement) bezeichnet wird. Da ein solcher Eingriff in den USA verboten war, wich der Arzt nach Mexiko aus, wo die Therapie weder ausdrücklich erlaubt noch verboten ist. Am 6. April 2016 wurde ein Junge geboren, der sich zumindest in den ersten Lebensmonaten normal entwickelte.
Der experimentell-therapeutische Ansatz
Naiv betrachtet könnte die Lösung des Problems darin bestehen, die mutierten Mitochondrien durch funktionsintakte Mitochondrien auszutauschen (mitochondrial replacement). Dass dies kaum praktikabel ist, erklärt sich bereits durch die große Zahl an Mitochondrien, die in einer Zelle enthalten sind. Konsequenterweise muss die alternative Option gewählt werden. Hierbei wird das Kerngenom der Eizelle in eine neue Umgebung transferiert, die funktionsintakte Mitochondrien enthält (nuclear genome transfer).
Vom Konzept her klingt das einfach und plausibel. Zunächst wird der Kern einer gesunden, anonymen Eizell-Spenderin aus der Eizelle entfernt und verworfen. Dann wird das Kern-Äquivalent einer Eizelle der künftigen Mutter in die entkernte Eizelle der Spenderin eingebracht.
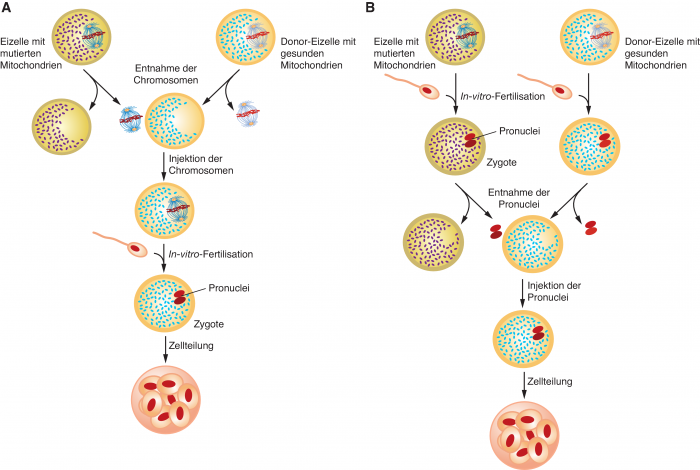
Dabei sind zwei Ansätze möglich (siehe Abb. 1):
- Bei einem maternalen Spindeltransfer (MST) wird der Zellkern im Stadium der Metaphase II der meiotischen Teilung mitsamt dem Spindelapparat aus der Eizelle der künftigen Mutter isoliert und in die entkernte Spender-Eizelle übertragen. Erst dann erfolgt die In-vitro-Befruchtung mit den Spermien des künftigen Vaters. Ein so erzeugter, gesund erscheinender Embryo wird dann der hormonell konditionierten künftigen Mutter eingesetzt, die den Fetus in einer normalen Schwangerschaft austrägt.
- Bei einem Vorkern-Transfer (Pronuclear-Transfer, PNT) findet der Austausch des Kerngenoms erst nach der Befruchtung statt. Hier werden die Pronuclei, also die Kerne des Spermas und der Eizelle noch vor ihrer Fusion aus einer frühen Zygote isoliert und in eine entkernte Zygote transferiert, die sich nach Befruchtung aus einer Spender-Eizelle mit dem Sperma des künftigen Vaters gebildet hat.
Beide Methoden unterscheiden sich also fundamental dadurch, dass einmal der Kerntransfer vor und einmal nach der Befruchtung stattfindet. Oder anders ausgedrückt: Beim maternalen Spindeltransfer wird eine Eizelle, beim Vorkern-Transfer ein Embryo manipuliert. Bei der jordanischen Frau wurde in Mexiko ein maternaler Spindeltransfer durchgeführt.
Anders lag der Fall bei einem Ehepaar in der Ukraine, das sich nach mehrfach missglückter In-vitro-Fertilisation ebenfalls zu einer Eizellspende entschlossen hatte, ohne dass irgendein genetischer Defekt in den Mitochondrien diagnostiziert worden war. Valeriy Zukin, der Direktor an der Nadiya Klinik in Kiew, entschloss sich zu einem Vorkern-Transfer bei dem Ehepaar und der Eizellspenderin – mit Erfolg: Anfang Januar kam ein gesunder Junge zur Welt.
Die Zukunft
Natürlich wird das Vorgehen des Therapeuten höchst kontrovers gesehen. Viele Kritiker glauben nachvollziehbar, dass die Mitochondrien-Spende noch nicht ausreichend getestet ist. Auch das erste, auf diese Art gezeugte Kind besitzt eine geringe Zahl mutierter Mitochondrien, die beim Spindel-Transfer mit übertragen wurden. Wie sich dies auf eine längere Sicht bei dem Kind auswirkt, bleibt abzuwarten.
Bemerkenswert ist auch, dass ein Junge geboren wurde. Man kann annehmen, dass dies kein Zufall ist, sondern dass sehr bewusst eine männliche Zygote der Mutter implantiert wurde. Denn so ist sichergestellt, dass es keine Nachkommen des Kindes geben wird, die auch einen Mosaik-Genotyp von drei Genomspendern haben werden, da ja nur Mütter Mitochondrien vererben.
Legal durchführen lässt sich das Verfahren derzeit nur in Großbritannien. Dort gilt die mitochondrial replacement therapy (MRT) nicht als genetischer Eingriff in die Keimbahn. In den USA hat eine Expertenkommission vorerst ein Abwarten empfohlen, dort gilt die MRT als genetische Manipulation der Keimbahnzellen – zumindest, wenn dabei weibliche Nachkommen entstehen. Und in Deutschland schließt die heutige Rechtslage eine Zulassung dieser Methode eindeutig aus.
So bleibt abzuwarten, welche Erfahrungen in Großbritannien mit der Mitochondrien-Spende gemacht werden. Erweist sich das Verfahren als sicher und erfolgreich, wird man sich auf Dauer wohl nicht dagegen aussprechen können, wenn tatsächlich eine eindeutige Indikation vorliegt und wenn sichergestellt ist, dass die Therapie von ausgewiesenen Experten durchgeführt wird. Betroffene werden diese Experten finden, und es ist gut so, dass ihnen geholfen werden kann. |
Literatur
[1] Amato P, Tachibana M, Sparman M, Mitalipov,S. Three-Parent IVF: Gene Replacement for the Prevention of Inherited Mitochondrial Diseases. Fertil Steril 2014;101:31–35
[2] Dimauro S, Davidzon G. Mitochondrial DNA and Disease. Annals of Medicine 2005;37:222–232
[3] Craven L, Elsom JL, Irving L, Harbottle SJ, Murphy JL, Cree LM et al. Mitochondrial DNA Disease: New Options for Prevention. Hum Mol Genet 2011;20:R168-R174
Autoren
Prof. Dr. Theo Dingermann ist Seniorprofessor am Institut für Pharmazeutische Biologie an der Goethe-Universität Frankfurt.
Dr. Ilse Zündorf ist dort als akademische Oberrätin tätig.
Institut für Pharmazeutische Biologie Biozentrum, Max-von-Laue-Straße 9 60438 Frankfurt/Main



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.