














- DAZ.online
- DAZ / AZ
- DAZ 29/2017
- Ähnlich, aber nicht ...

Foto: chechotkin – Fotolia.com
Zulassung
Ähnlich, aber nicht gleich
Vereinfachtes Zulassungsverfahren von Enoxaparin-Biosimilars birgt Risiken
Niedermolekulare Heparine (NMH) werden aus unfraktioniertem Heparin (UFH), das im Wesentlichen aus Schweinedärmen isoliert wird, gewonnen. Sie bieten – je nach Präparat – gegenüber UFH eine Reihe von Vorteilen wie
- die dosisunabhängig höhere absolute Bioverfügbarkeit (≥ 90%) nach s.c. Gabe,
- die Möglichkeit der s.c. Einmalgabe pro Tag zur Prophylaxe und Therapie venöser Thromboembolien (VTE),
- den möglichen Verzicht auf ein Routine-Monitoring,
- die bessere Verträglichkeit (z. B. geringeres Osteoporose-Risiko) sowie
- eine höhere Sicherheit (z. B. geringere Immunogenität im Sinne einer Heparin-induzierten Thrombozytopenie Typ II).
Darüber hinaus haben klinische Studien in bestimmten Indikationen Vorteile von NMH gegenüber UFH gezeigt (z. B. eine geringere Inzidenz an rezidivierenden Thrombosen bei Tumorpatienten mit VTE) [1].
Vorgaben des Europäischen Arzneibuchs
Gemäß Ph. Eur. 8 müssen Niedermolekulare Heparine (Heparina massae molecularis minoris) eine mittlere Molekülmasse unter 8000 Da aufweisen, wobei mindestens 60% (m/m) eine relative Molekülmasse von weniger als 8000 haben sollen. Die Anti-Faktor-Xa-Aktivität (aXa-Aktivität) muss mindestens 70 IE pro Milligramm, der Quotient der aXa-Aktivität zur Anti-Faktor-IIa-Aktivität (aIIa-Aktivität) mindestens 1,5 betragen.
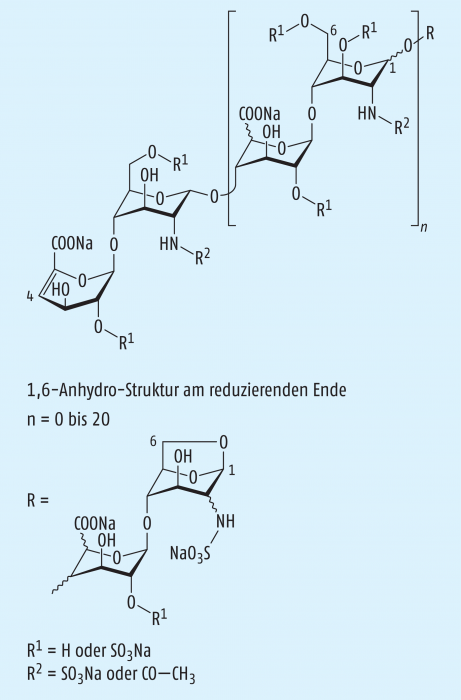
Beim Enoxaparin-Natrium, das durch alkalische Depolymerisation der Benzylester-Derivate aus UFH gewonnen wird, handelt es sich um ein komplexes Gemisch von Oligosacchariden, das noch nicht vollständig in allen Bestandteilen identifiziert werden konnte. Der Hauptteil der Komponenten weist eine 4-Enopyranoseuronat-Struktur am nicht-reduzierenden Ende der Kette auf, und 15 bis 25% der Komponenten besitzen am reduzierenden Ende der Kette eine 1,6-Anhydro-Struktur (Abb. 1). Die mittlere Molekülmasse liegt im Bereich von 3800 bis 5000 Da, wobei der charakteristische Mittelwert etwa 4500 Da beträgt. Der Grad der Sulfatierung je Disaccharid-Einheit liegt bei etwa 2. Die aXa-Aktivität beträgt mindestens 90 IE und höchstens 125 IE je Milligramm, die aIIa-Aktivität mindestens 20 IE und höchstens 35 IE je Milligramm, jeweils berechnet auf die getrocknete Substanz. Der aXa/aIIa-Quotient liegt zwischen 3,3 und 5,3.
Bei der Identitätsprüfung unter der Verwendung von Enoxaparin-Natrium CRS wird vorausgesetzt, dass die Heparinketten mit einer relativen Molekülmasse < 2000 Da einen Anteil von 12,0 bis 20,0% (m/m) und diejenigen zwischen 2000 und 8000 Da einen Anteil von 68,0 bis 82,0% haben. Zur Reinheitsprüfung sieht die Monografie vor: Messungen zum Aussehen der Lösung (klar, nur mäßig gefärbt), des pH-Werts (6,2 – 7,7), von Benzylalkohol (höchstens 0,1% m/m) und Natrium (11,3 – 13,5%) [2].
Präparatespezifische Produktmuster
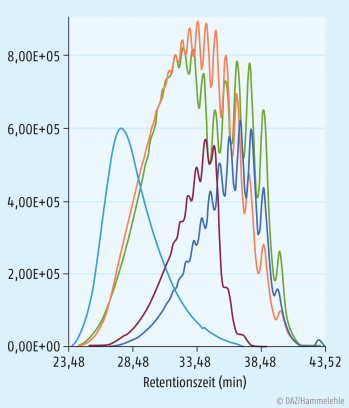
Die jeweiligen Verfahren zur NMH-Gewinnung führen zu nicht identischen Endprodukten, wie einige Parameter zeigen, z. B. durchschnittliche Molekülmasse, Streubereich höher- und niedermolekularer Fraktionen, aXa/aIIa-Quotient (s. Tab. 1 und Abb. 2) [3].
Wirkstoff |
Handelsname (Beispiele) |
Indikationen |
Mittlere Molekülmasse |
aXa/aIIa-Quotient |
aXa t1/2
|
Dosierung Prophylaxe (IE aXa/Tag) |
Dosierung VTE-Therapie (IE aXa/Tag) |
|---|---|---|---|---|---|---|---|
Certoparin |
Monoembolex |
A – D |
4,2 – 6,2 kDa |
ca. 2,2 : 1 |
4,3 h |
A – C: 3000 IE |
2-mal 8000 IE |
Dalteparin |
Fragmin |
A – E, G |
ca. 6 kDa |
ca. 2,5 : 1 |
3 – 5 h |
A, C: 5000 IE
B: 2500 IE
|
1-mal 200 IE/kg KG oder 2-mal 100 IE/kg KG |
Enoxaparin |
Clexane |
A – F |
ca. 4,5 kDa |
3,3 – 5,3 : 1 |
4,5 h |
B: 2000 IE
A, C: 4000 IE
|
2-mal 100 IE/kg KG
1-mal 150 IE/kg KG
|
Nadroparin |
Fraxiparin, Fraxodi |
A, B, D, E |
4,9 kDa |
ca. 4 : 1 |
3,3 h |
A: nach KG-Klassen
B: 2850 IE
|
2-mal 87,5 IE/kg KG oder 1-mal 175 IE/kg KG |
Reviparin |
Clivarin |
A, B, D |
4,7 kDa |
3,6 – 6,1 : 1 |
3,0 h |
A: 3436 IE
B: 1750 IE
|
2-mal 2863 IE (35 – 45 kg KG)
2-mal 3436 IE (46 – 60 kg KG)
2-mal 5135 IE (> 60 kg KG)
|
Tinzaparin |
Innohep |
A – E, G |
ca. 6,5 kDa |
1,5 – 2,5 : 1 |
3,3 h |
B: 3500 IE
A, C: 4500 IE
|
1-mal 175 IE/kg KG |
VTE = venöse Thromboembolien; aIIa = Anti-Faktor-IIa-Aktivität; aXa = Anti-Faktor-Xa-Aktivität; t1/2 = Eliminationshalbwertszeit. | |||||||
Die pharmakologische Aktivität der NMH beschränkt sich nicht auf die aXa- und aIIa-Aktivität, sondern beruht auf weiteren, „pleiotropen“ Effekten wie der Freisetzung des Tissue-Factor-Pathway-Inhibitors (TFPI) aus dem Endothel und der Hemmung der Fibrinopeptid-A-Bildung sowie auf antiinflammatorischen (z. B. Blockade der Selectin-Freisetzung) und antiangiogenetischen Mechanismen (z. B. Anstieg der TGF-β1 -Konzentration) [4].
Wegen der unterschiedlichen physikochemischen und klinisch-pharmakologischen Eigenschaften war es für jeden NMH-Hersteller unumgänglich, für jede angestrebte Indikation seines Präparates eine klinische Studie vorzulegen, um eine entsprechende Zulassung zu erwirken. Das Spektrum der zugelassenen Indikationen bei den verschiedenen NMH ist bis heute nicht einheitlich, wie z. B. die Therapie von Herzinfarkten zeigt – was an fehlenden Studien oder einer nicht adäquaten Wirkung oder Sicherheit des geprüften NMH liegen kann.
Glomeruläre Filtrationsrate beachten!
Insbesondere bei geriatrischen Patienten, die mit einem NMH behandelt werden, ist zur richtigen Dosierung eine möglichst genaue GFR-Bestimmung erforderlich. Wenn die GFR nach Cockcroft-Gault oder MDRD berechnet wird, kann es allerdings leicht zu einer Überschätzung oder Verzerrung der wahren GFR kommen, sodass es sinnvoll erscheint, bei nicht-onkologischen geriatrischen Patienten vorzugsweise auf eine Cystatin-C-basierte Formel zurückzugreifen (z. B. Cystatin-C-basierte CKD-EPI-Formel) [7].
Enoxaparin: s.c. Einmalgabe pro Tag bei VTE
Da das NMH Enoxaparin (Clexane®) über nationale Zulassungsverfahren sukzessive in verschiedenen europäischen Ländern zugelassen wurde, gab es länderspezifische Abweichungen in den Zulassungstexten. Am 15. Dezember 2016 hat die EMA eine Empfehlung für die Produktinformationen für Clexane® mit harmonisierten Wirksamkeits- und Sicherheitsangaben für alle Länder der EU ausgesprochen. Mit der EU-weiten Harmonisierung der Produktinformationen (Verfahren nach Artikel 30 der Richtlinie 2001/83/EG, Referenznummer EMEA/H/A-30/1429) wurden diese nationalen Zulassungen in Zulassungen nach dem Verfahren der gegenseitigen Anerkennung (Mutual Recognition Procedure, MRP) transferiert.
So steht beispielsweise in einigen europäischen Ländern schon seit vielen Jahren in der Fachinformation des Enoxaparin-haltigen Fertigarzneimittels, dass es bei manifester venöser Thromboembolie (VTE) auch einmal täglich mit einer Dosierung von 1,5 mg/kg s.c. verabreicht werden kann, während diese Dosierung in Deutschland bis vor Kurzem nicht zugelassen war. Allerdings setzt die Einmalgabe eine Risikostratifizierung des Patienten voraus (Tab. 2).
Diese Zulassung beruht auf den Ergebnissen einer Studie, in der zwei Dosierungen von Enoxaparin – d. h. einmal täglich 1,5 mg/kg s.c. versus zweimal täglich 1 mg/kg s.c. – bei manifester VTE im Vergleich zu UFH geprüft worden waren. Dabei waren keine signifikanten Unterschiede in der klinischen Wirksamkeit und Sicherheit zwischen den beiden Enoxaparin-Anwendungen zu erkennen gewesen [5].
Im Rahmen dieses EU-Harmonisierungsprozesses sind noch weitere Angleichungen vorgenommen worden, die zukünftig zu berücksichtigen sind (Tab. 2).
Indikation |
vor der Harmonisierung |
Stand März 2017 |
Kommentar |
|---|---|---|---|
Prophylaxe bei mäßigem Risiko für Thromboembolien |
1 × 2.000 IE (20 mg) 2 Std. vor Operation und täglich nach Operation |
wie vorher |
Die frühere Formulierung „niedriges und mittleres Risiko“ wurde in „mäßiges Risiko“ geändert. |
Prophylaxe bei mäßigem und hohem Risiko für Thromboembolien bei GFR 15 – 30 ml/min |
niedriges und mittleres Risiko: 1 × 2.000 IE
hohes Risiko: 1 × 3000 IE
|
mäßiges Risiko: 1 × 2000 IE
hohes Risiko: 1 × 2000 IE
|
Die Dosis wurde unabhängig vom Risiko vereinheitlicht. |
Therapie venöser Thromboembolien (VTE) bei GFR ≥ 30 ml/min* |
unabhängig vom Komplikationsrisiko:
2 × 1 mg/kg/Tag s.c.
|
geringes Risiko für erneute VTE-Ereignisse: 1 × 1,5 mg/kg/Tag s.c.
erhöhtes Risiko für Blutungen und erneute VTE-Ereignisse: 2 × 1 mg/kg/Tag s.c.
|
Die neue Dosierungsempfehlung setzt eine Risikoklassifizierung des Patienten voraus. Erhöht ist das Risiko z. B. bei Adipositas, symptomatischer Lungenembolie, proximaler Thrombose (Vena iliaca), Tumorerkrankung oder rezidivierender VTE. |
Prävention extrakorporaler Thromben während der Hämodialyse |
Standard: 1 mg/kg (zu Beginn der Sitzung); je nach Blutungsrisiko und Gefäßzugang: 50–75 IE kg/KG |
wie vorher |
|
* Bei GFR 15–30 ml/min: weiterhin 1 × 1 mg/kg/Tag s.c. | |||
Tinzaparin: Thromboseprophylaxe im Hochrisikobereich
Tinzaparin (Innohep®) unterscheidet sich in seinen physikochemischen Eigenschaften deutlich von Enoxaparin. Seine halbsynthetische Gewinnung aus unfraktioniertem Heparin (UFH) mittels Heparinase führt zu einem höheren Anteil an höhermolekularen polysulfatierten Polysaccharid-Einheiten. Daher ist nicht nur die durchschnittliche Molekülmasse deutlich höher als beim Enoxaparin, sondern auch der aXa/aIIa-Quotient geringer (Tab. 1), denn die aXa-Aktivität geht primär von den niedermolekularen Fraktionen aus, die aIIa-Aktivität hingegen von höhermolekularen Anteilen. Zudem wird beim Tinzaparin ein geringerer Anteil der verabreichten Dosis in aktiver Form über die Nieren ausgeschieden [6].
Auch die Fachinformation von Innohep® wurde EU-weit harmonisiert, nachdem das Präparat beispielsweise schon seit Jahren in Skandinavien für die Thromboembolieprophylaxe bei Hochrisikopatienten zugelassen war (sowohl bei chirurgischen Eingriffen, z. B. Hüft- und Kniegelenksersatz oder Resektion von Tumoren, als auch bei immobilisierten Patienten), während es in Deutschland bis vor Kurzem nur für Eingriffe mit mittlerem Thromboembolie-Risiko einsetzbar war. Diese EU-Harmonisierung ist auch deshalb nachvollziehbar, weil schon vor mehr als 20 Jahren eine Vergleichsstudie zwischen Enoxaparin 4000 IE/Tag und Tinzaparin 4500 IE/Tag zur Prophylaxe bei orthopädischen Eingriffen (Hüftgelenksersatz) keinen signifikanten Unterschied in der klinischen Wirksamkeit und Verträglichkeit zwischen beiden Präparaten ergab [8].
Vereinfachte Zulassung von Enoxaparin-Biosimilars
Schon vor mehr als zehn Jahren wurde geregelt, dass in Europa die Zulassung eines niedermolekularen Heparins (NMH) durch einen Zweitanbieter nicht auf nationaler Ebene, sondern ausschließlich über die Europäische Zulassungsbehörde EMA erfolgen kann. Damals wurde auf der Basis der Richtlinie für ähnliche biologische Arzneimittel (CHMP/437/04) eine produktspezifische Klassifikation für NMH implementiert. Im vierten Quartal 2016 hat die EMA die beiden Enoxaparin-Biosimilars Inhixa® und Thorinane® zugelassen, die allerdings bisher nicht in Deutschland verfügbar sind. Die beiden Zulassungs- bzw. Lizenzinhaber, die Firmen Techdow Europe AB und Pharmathen S.A., sind im Biosimilar-Markt bisher nicht in Erscheinung getreten.
Überraschend ist, dass die EMA in den Zulassungsverfahren von Inhixa® und Thorinane® deutliche Abweichungen zu den bisher europaweit zugelassenen Biosimilars erlaubt hat, die bei Experten mehrere Fragen aufwerfen [18].
Bei den bisher zugelassenen Biosimilars handelte es sich durchweg um rekombinant hergestellte Proteine oder Glykoproteine. Der Biosimilar-Hersteller musste nicht nur sehr umfangreiche Daten zu den physikochemischen Eigenschaften seines Produkts, sondern auch Studienergebnisse zur Bioäquivalenz an gesunden Probanden vorlegen sowie in einer Phase-III-konformen Vergleichsstudie die Nicht-Unterlegenheit gegenüber dem Originalprodukt hinsichtlich der klinischen Wirksamkeit in der definierten Indikation, Verträglichkeit und Sicherheit (z. B. potenzielle Immunogenität im Follow-up) belegen.
Dieses Prozedere der EMA erfährt weltweit große Anerkennung, da es bis zum jetzigen Zeitpunkt keinen Anlass gab, die Qualität der zugelassenen Produkte infrage zu stellen. Selbst die Tatsache, dass der Biosimilar-Hersteller die Chance hat, mit den positiven Ergebnissen einer Phase-III-konformen Studie in einer bestimmten Anwendung die Zulassung für weitere Indikationsgebiete zu erhalten (Extrapolation), wird nach anfänglichen Vorbehalten inzwischen von Fachkreisen weitgehend akzeptiert [9].
Bei den Zulassungsverfahren der Enoxaparin-Biosimilars Inhixa® und Thorinane® sind allerdings deutliche Abweichungen zum bisherigen Prozedere für Biosimilars rekombinanten Ursprungs zu erkennen: Die Antragsteller mussten keine Phase-III-konforme Vergleichsstudie in einer bestimmten Indikation vorlegen, sondern neben der physikochemischen Charakterisierung gemäß Ph. Eur. 8 und einer Reihe präklinischer Daten waren vergleichende Untersuchungen zur aXa-, aIIa- und TFPI-Aktivität bei gesunden Probanden für die Zulassung ausreichend [10].
Kritiker dieses vereinfachten Zulassungsverfahrens argumentieren, dass es sich bei einem NMH um kein definiertes Protein, sondern um ein komplexes Gemisch von Oligosacchariden biologischen Ursprungs handelt, das bis heute noch nicht vollständig in seinen Bestandteilen aufgeklärt ist (s.o.). Zudem ist die Inzidenz möglicher Überempfindlichkeitsreaktionen Typ IV und pseudoallergischer Reaktionen fraglich, sodass letztendlich nur Meldungen im Rahmen der Pharmakovigilanz mehr Klarheit über die tatsächliche Sicherheit der Enoxaparin-Biosimilars bringen werden [11].
Risiken von niedermolekularen Heparinen
Heparin-induzierte Thrombozytopenie (HIT) Typ II. Bei der HIT Typ II handelt es sich um eine sehr ernstzunehmende Überempfindlichkeitsreaktion auf Heparin(-derivate), vor allem wenn Immunglobulin-vermittelt Quervernetzungen von Thrombozyten entstehen, die sowohl zu einem messbaren Abfall der Thrombozytenkonzentration als auch zu lebensbedrohlichen arteriellen Thromboembolien führen, wenn nicht rechtzeitig interveniert wird [12]. Während die Hersteller von NMH-Originalpräparaten mittlerweile umfangreiche Daten zur Sicherheit bezüglich HIT Typ II vorlegen können, ist dies bei den Biosimilar-Herstellern nicht der Fall.
Potenzielle Immunogenität. Bekannt ist seit Langem, dass Heparin-PF-4-Komplexe (PF-4 = platelet factor 4) eine Antikörper-Antwort und die Aktivierung dendritischer Zellen hervorrufen können. Bis heute existiert jedoch noch kein Test, mit dem man sicher das immunogene Potenzial von Heparin-Präparaten nachweisen kann.
Eine Studiengruppe in den USA verwendete einen MIMIC® PTE-Assay (Peripheral Tissue Equivalent), um die Sicherheit von Enoxaparin-Biosimilars zu testen. Dabei zeigten sich nicht nur Abweichungen in der Freisetzung des Tissue-Factor-Pathway-Inhibitors (TFPI) gegenüber dem Originalpräparat, sondern auch Anzeichen einer erhöhten immunogenen Potenz. Die Autoren machten dafür zum einen Spuren von hochmolekularen polysulfatierten Polysacchariden – wie unfraktioniertes Heparin (UFH) – zum anderen physikochemisch stabilere NMH-PF-4-Komplexe verantwortlich. Besonders nachdenklich stimmt die Beobachtung, dass bei den Biosimilars – im Gegensatz zum Originalpräparat – von Charge zu Charge deutliche Schwankungen der TFPI-Freisetzung nachweisbar waren, was die Konformität des halbsynthetischen Herstellungsverfahrens infrage stellt. Dies ist auch deshalb interessant, weil möglicherweise eine inverse Korrelation zwischen TFPI-Freisetzung und Immunogenität besteht [13].
Niedermolekulare Heparine versus NOAK
Stieg in den 1990er-Jahren der NMH-Einsatz weltweit kontinuierlich an, so stagniert er inzwischen, was teilweise mit der Markteinführung der neuen oralen Antikoagulanzien (NOAK) zusammenhängt. Allerdings sind die NOAK bis heute weder für die Thromboembolie-Prophylaxe bei Dialysepatienten noch für internistische Indikationen noch für Schwangere zugelassen. Darüber hinaus liegen bisher keine ausreichenden Studiendaten für ihren Einsatz bei Tumorpatienten mit akuter tiefer Beinvenenthrombose oder Lungenembolie vor. Selbst in orthopädischen Zentren werden weiterhin sehr häufig NMH zur präoperativen Thromboembolieprophylaxe verwendet, da sie eine absolute Bioverfügbarkeit ≥ 90% haben und die postoperative Wundheilung erfahrungsgemäß nicht beeinträchtigen.
Diskussion
Es ist bemerkenswert, dass hämatologische und hämostaseologische Experten in Australien bereits 2014 ein Konsensuspapier auf den Weg gebracht haben, um vor einer möglichen Zulassung von NMH-Biosimilars deren Stellenwert zu klassifizieren [11]. Aufgrund der komplexen Struktur der NMH halten sie es für praktisch unmöglich, dass ein NMH-Biosimilar mit dem Originalpräparat identisch ist, was auch die antikoagulatorische Aktivität und die potenzielle Immunogenität betrifft. Aus ihrer Sicht ist eine breite klinische Anwendung eines NMH-Biosimilars erst dann möglich, wenn seine Wirksamkeit und Sicherheit zuvor in einer prospektiven Studie im Vergleich mit dem Originalpräparat geprüft wurde und wenn die Anwendung von einem Postmarketing-Pharmakovigilanz-Programm begleitet wird.
In Bioäquivalenz-Untersuchungen ist es nicht möglich, NMH-Plasmakonzentrationen direkt zu messen, sodass indirekte Parameter herangezogen werden müssen, die neben der Anti-Xa- oder Anti-IIa-Aktivität die pleiotropen Effekte der NMH abbilden. Bemerkenswert ist in diesem Zusammenhang die Tatsache, dass bei den Untersuchungen zur TFPI-Aktivität von Inhixa® die AUC (IE ⋅ h/ml) nur bei 85,10% des Originalpräparates lag (95%-Konfidenzintervall 56,46 – 128,29%), was Fragen zur Bioäquivalenz aufwirft [10].
Bei biotechnologisch (d. h. rekombinant) hergestellten Arzneimitteln ist zum gegenwärtigen Zeitpunkt ein Aut-idem-Austausch in Deutschland nicht vorgesehen, wenn der jeweilige Patient mit dem Originalpräparat anbehandelt ist. Im Rahmenvertrag über die Arzneimittelversorgung (gemäß § 129 Abs. 1a SGB V) existiert aber bisher keine Regelung zur Aut-idem-Substitution mit Biosimilars, die nicht-biotechnologisch hergestellt sind. Diese Regelungslücke sollte unter den dargestellten Vorzeichen zeitnah geschlossen werden. Alternativ könnte Enoxaparin bis auf Weiteres in die Substitutionsausschlussliste aufgenommen werden.
Der potenzielle klinische Stellenwert eines Biosimilars lässt sich u. a. mittels SOJA (system of objectified judgement analysis) beurteilen (Tab. 3) [15]. Im Fall der Enoxaparin-Biosimilars würde die Summe aufgrund mehrerer fehlender klinischer Anhaltspunkte derzeit deutlich unter 1000 Punkten liegen. Weitergehende, modifizierte SOJA-Analysen berücksichtigen auch die durchgängige Lieferfähigkeit des Unternehmens, um einen Präparatewechsel zu vermeiden. Beide Unternehmen, Techdow und Pharmathen, lassen derzeit keine entsprechende Bewertung zu.
Kriterium |
Punktezahl |
|---|---|
Anzahl der Formulierungen |
60 |
Anzahl der zugelassenen Indikationen |
20 |
Variabilität der AUC |
35 |
Arzneimittelinteraktionen |
80 |
klinische Wirksamkeit |
290 |
Nebenwirkungen |
215 |
Dosierungshäufigkeit |
120 |
Kosten (stationär/ambulant) |
90 |
Dokumentation* |
90 |
Summe |
1000 |
* Anzahl der weltweit behandelten Patienten und Erfahrungen zur Pharmakovigilanz | |
Es ist unklar, weshalb die EMA bei Enoxaparin das bisher erfolgreiche Zulassungsverfahren für Biosimilars geändert hat, obwohl hier ein komplexes, bisher nicht eindeutig definiertes Gemisch biogenen Ursprungs vorliegt, bei dem verschiedene pleiotrope Effekte wahrscheinlich eine substanzielle Rolle spielen [16, 17].
Für die in gewisser Weise vergleichbaren intravenös applizierbaren Eisen(III)-Kohlenhydrat-Komplexen fordern Experten inzwischen stringentere zentrale Zulassungsverfahren, da Unsicherheiten zu ihrer klinischen Wirksamkeit und Verträglichkeit bestehen, die wahrscheinlich den vergleichsweise einfachen nationalen Zulassungsverfahren geschuldet sind [18]. NMH sind hinsichtlich ihrer pharmazeutischen Äquivalenz und ihrer Bioäquivalenz ähnlich herausfordernd wie die Eisen(III)-Kohlenhydrat-Komplexe.
Fazit
Der EU-Harmonisierungsprozess von NMH-Originalpräparaten wie Clexane® oder Innohep® ist nachvollziehbar und durch entsprechende Phase-III-Studien abgedeckt. Hingegen wirft die zentrale Zulassung der Enoxaparin-Biosimilars Inhixa® und Thorinane® Fragen zur klinischen Wirksamkeit und Sicherheit auf, solange keine weitergehenden überzeugenden Studiendaten vorliegen. |
Literatur
[1] Jeske WP et al. Differentiating Low-Molecular-Weight Heparins Based on Chemical, Biological, and Pharmacologic Properties: Implications for the Development of Generic Versions of Low-Molecular-Weight Heparins. Semin Thromb Hemost 2008;34:74-85
[2] Monografie Enoxaparin-Natrium. Ph. Eur. 8. Ausgabe, 1. Nachtrag, 2014
[3] Mulloy B et al. Molecular Weight Measurements of Low Molecular Weight Heparins by Gel permeation Chromatography. J Thromb Haemost 1997;77:668-674
[4] Walenga JM, Jackson CM, Kessler CM. Low molecular weight heparins differ substantially: Impact on developing Biosimilar drugs. Semin Thromb Hemost 2011;37:322-327
[5] Merli G et al. Subcutaneous enoxaparin once or twice daily compared with intravenous unfractionated heparin for treatment of venous thromboembolic disease. Ann Intern Med 2001;134:191-202
[6] McCart GM, Kayser SR. Therapeutic Equivalency of Low-Molecular-Weight Heparins. Ann Pharmacother 2002;36:1042-1057
[7] Schäffner E. Bestimmung der Glomerulären Filtrationsrate. Klinikarzt 2017;46:74-78
[8] Planes A et al. Prevention of deep vein thrombosis after hip replacement – Comparison between two low-molecular heparins, tinzaparin and enoxaparin. J Thromb Haemost 1999;81:22-25
[9] Lipp H-P. Biosimilars mit EU-Zulassung! Was kann heute schon therapeutisch genutzt werden? Kompendium Biosimilars 2016;1:33-38
[10] EMA. Inhixa; www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004264/human_med_002020.jsp&mid=WC0b01ac058001d124; Stand 25.4.2017
[11] Nandurkar H et al. Australian Low-Molecular-Weight Heparin Biosimilar Working Group (ALBW). Low-molecuar-weight heparin biosimilars: potential implications for clinical practice. Intern Med J 2014;44:497-500
[12] Caren G, Solomon M. Heparin-Induced Thrombocytopenia. N Engl J Med 2015;373:252-261
[13] Luna E et al. Evaluation of Immunostimulatory Potential of Branded and US-Generic Enoxaparins in an In Vitro Human Immune System Model. Clin Appl Thromb Hemost 2015;21:211-222
[14] Chao J et al. Nomenclature and Traceability Debate for Biosimilars: Small-Molecule Surrogates Lend Support for Distinguishable Nonproprietary Names. Adv Ther 2015;32:270-283
[15] Janknegt R, Scott M, Mairs J. System of Objectified Judgement Analysis (SOJA) as a tool in rational and transparent drug-decision making. Expert Opin Pharmacother 2007;8(Suppl 1):5-14
[16] Walenga JM et al. Comparative Studies on branded enoxaparin and a US generic version of enoxaparin. Clin Appl Thromb Hemost 2013;19:261-267
[17] Jeske W et al. Update on the safety and bioequivalence of biosimilars – focus on enoxaparin. Drug Healthcare Patient Safety 2014;5:133-141
[18] Hussaarts L et al. Equivalence of complex drug products: advances in and challenges for current regulatory frameworks. Ann NY Acad Sci; Epub 26.4.2017
Autor
Prof. Dr. rer. nat. Hans-Peter Lipp, Universitätsapotheke Tübingen



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.