


















- DAZ.online
- DAZ / AZ
- DAZ 18/2017
- Der Feind im eigenen Kö...

Foto: pressmaster – Fotolia.com
Immunologie
Der Feind im eigenen Körper
Wie Autoimmunerkrankungen entstehen
Hypersensitivität und Allergie sowie – bei Patienten nach einer Organtransplantation – eventuell eine Abstoßungsreaktion treten sehr wohl auf. Eskaliert das Immunsystem und richtet sich sogar gegen körpereigene Substanzen, resultieren Autoimmunerkrankungen. Immerhin 5 bis 8% der Bevölkerung in Deutschland leiden an einer Autoimmunerkrankung, und ungefähr 20% sind von einer Allergie betroffen – in beiden Fällen ist die Tendenz steigend.
Wie kann es sein, dass das derart ausgeklügelte Immunsystem so aus der Kontrolle gerät, dass es für den Organismus zur Belastung bis hin zur Lebensbedrohung wird? Um das zu verstehen, müssen wir uns mit einem zentralen Begriff auseinandersetzen: „Toleranz“! Toleranz bedeutet, dass unser Immunsystem lernen muss, nicht gegen die körpereigenen Strukturen aktiv zu werden.
Vorweg drei Vorbemerkungen:
- Toleranz ist ausschließlich ein Problem des adaptiven (spezifischen) Immunsystems.
- Für Selbsttoleranz ist keinerlei Information in den Genen für die wichtigen Antigen-Erkennungsmoleküle des spezifischen Immunsystems, die T- und B-Zell-Rezeptoren, abgelegt.
- Weil T- und B-Zell-Rezeptoren zunächst unabhängig von einem Antigenkontakt durch einen zufallsgesteuerten Prozess entstehen, muss eine Autoreaktivität durch Prozesse verhindert werden, die zu einer Toleranz gegen körpereigene Strukturen führen.
Zudem sei vorweggeschickt, dass sich immunologische Toleranz darin äußert, dass das Immunsystem gegen bestimmte Moleküle inaktiv bleibt, obwohl diese Moleküle vom Immunsystem erkannt werden. Daraus folgt, dass bei der Toleranz die Effektorphase des spezifischen Immunsystems betroffen ist.
Aus überschaubarer Information wird unglaubliche Vielfalt
Bezogen auf die enorme molekulare Heterogenität unseres adaptiven Immunsystems, die sicherstellt, dass praktisch jedes mögliche Antigen erkannt werden kann, ist die Genausstattung für die Antikörper und die T-Zell-Rezeptoren als die wichtigsten Erkennungs- und Eliminationssysteme in unserem Immunsystem sehr übersichtlich. Schließlich wissen wir aus den Ergebnissen des humanen Genomprojektes, dass wir Menschen gerade einmal über ca. 20.000 Gene verfügen, was absolut nicht ausreichend ist, damit ca. 1012 verschiedene Antikörper und 1013 unterschiedliche T-Zell-Rezeptoren codiert werden. Die Vielfalt dieser Moleküle kommt durch zwei prinzipielle Mechanismen auf Einzelzellebene zustande, die normalerweise in unseren Körperzellen unbedingt auszuschließen sind. Das ist zum einen die Neukombination von Gensegmenten und zum anderen eine erstaunliche Ungenauigkeit bei Reparaturmechanismen im Rahmen dieser genomischen Neukombination. Dies alles hat zwei Konsequenzen:
- Es werden unglaublich viele spezifische Strukturen gebildet, die in ihrer Gesamtheit praktisch jede beliebige molekulare Oberfläche erkennen können.
- Diese unterschiedlichen Strukturen können auf Fremdmolekülen, aber auch auf körpereigenen Molekülen vorliegen. Alles wird gemacht, was theoretisch denkbar ist.
Das legt nahe, dass es Mechanismen geben muss, die diejenigen Moleküle aussortieren, die in der Lage wären, körpereigene Strukturen anzugreifen. Diese Mechanismen führen zu dem, was wir als Toleranz bezeichnen.
Zentrale versus periphere Toleranz
Prinzipiell lassen sich zwei Typen von Toleranz unterscheiden:
- Die zentrale Toleranz: Sie entwickelt sich im Rahmen der Reifung von T- und B-Zellen in den zentralen lymphatischen Organen, dem Knochenmark und dem Thymus.
- Die periphere Toleranz: Sie greift auf einer Ebene, wo sich die Immunzellen bereits zur Reife entwickelt haben.
Für die Entstehung und Aufrechterhaltung der immunologischen Toleranz ist die ständige Anwesenheit der Antigene notwendig, gegen die eine Toleranz entstehen soll. Diese Antigene werden auch als Tolerogene bezeichnet.
Toleranz bei B-Zellen. Für die Antikörper findet der wichtige Aussortierungsprozess, der zunächst die „zentrale Toleranz“ vermittelt, auf der frühen Ebene der B-Zell-Entwicklung im Knochenmark statt. Erst wenn sowohl die Genbausteine für die schwere als auch die für die leichte Antikörper-Kette funktional zusammengefügt sind, wird ein entsprechendes IgM-Molekül als B-Zell-Rezeptor auf der Oberfläche der unreifen B-Zelle exprimiert. Auf dieser Stufe findet der erste Kontakt mit denjenigen Selbstantigenen statt, die von Stroma-Zellen in der näheren Umgebung gebildet werden. Prinzipiell kann die Toleranz durch vier verschiedene Mechanismen etabliert werden (Abb. 1):
- Zelltod durch Apoptose oder klonale Deletion, vor allem durch polyvalente Autoantigene, da die autoreaktiven B-Zellen keine anti-apoptotischen Proteine (z. B. Bcl-2) exprimieren.
- Alternativ kann ein polyvalentes Autoantigen die Synthese eines neuen Rezeptors durch „Rezeptor-Editing“ der leichten Ketten induzieren. Das erste rearrangierte Gen für die leichte Kette wird verworfen und das zweite Allel neu zusammengebaut, wodurch sich die Rezeptorspezifität ändert.
- Induktion einer permanenten funktionellen Unempfindlichkeit gegenüber einem löslichen (monovalenten) Autoantigen, was auch als Anergie bezeichnet wird. Solche Zellen wandern in die Peripherie aus, regulieren aber die Expression von Oberflächen-IgM herunter und behalten nur den IgD-Antigenrezeptor.
- Immunologische Ignoranz: B-Zellen mit einer niedrigen Affinität zu einem löslichen monovalenten Autoantigen sind nicht zwingend inaktiv. Sie wandern in die Peripherie aus, wo sie unter normalen Bedingungen nicht stören, da dort die Konzentration des Autoantigens für eine Aktivierung konstant zu niedrig ist. Das kann sich aber unter bestimmten pathologischen Situationen durchaus ändern.
Im Idealfall sollten daher nur B-Zellen das Knochenmark verlassen, die keine körpereigenen Strukturen erkennen. Diese wandern dann in die peripheren lymphoiden Gewebe ein, wo sie zu Zellen reifen, die bei Bedarf einen spezifischen Antikörper in Massen produzieren. Insgesamt geht man davon aus, dass 85% der neu gebildeten B-Zellen im Knochenmark ausgemustert werden und nur 15% über das Blut in die Peripherie auswandern.
In der Milz findet anschließend der nächste Selektions- und Reifungsprozess statt, so dass die Zahl der potenziell autoreaktiven B-Zellen nochmals reduziert wird.
Schließlich bilden wahrscheinlich nur ca. 5 bis 10% der ursprünglich bereitgestellten B-Zellen einen Pool reifer, naiver B-Lymphozyten. Sie stehen bereit für eine Stimulation durch ein passendes Antigen und der entsprechenden T-Helferzelle und differenzieren dann zu antikörperproduzierenden Plasmazellen und B-Gedächtniszellen – der ganz normale Vorgang zur Pathogenabwehr. In diesem Stadium kann eine B-Zell-Toleranz auch als Konsequenz der T-Zell-Toleranz entstehen, da autoreaktive B-Zellen kein Aktivierungssignal einer passenden T-Helferzelle bekommen.
Toleranz bei T-Zellen. Auf ähnliche Weise wie die B-Zellen durchlaufen auch T-Zellen einen komplexen Selektionsmechanismus, der einen positiven und einen negativen Ast besitzt und der für viele Lymphozyten den Tod durch Apoptose bedeutet.
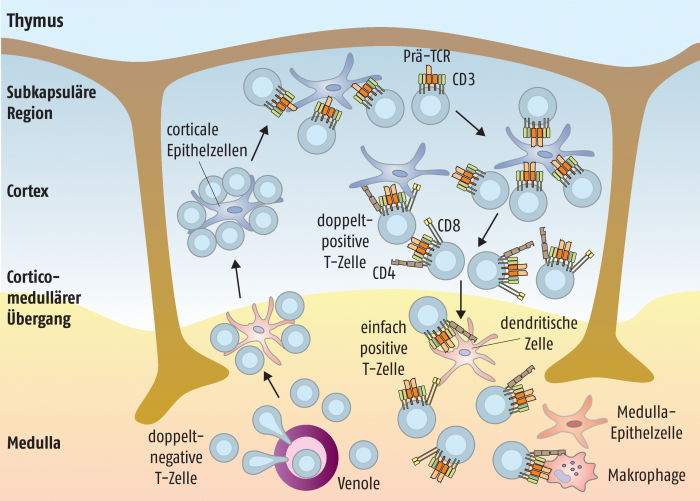
Der Reifungsprozess erfolgt im Thymus, in den die Vorläufer-T-Zellen, aus dem Knochenmark kommend, eingewandert sind. Diese Vorläufer-Zellen tragen noch keine der typischen Oberflächenproteine. Erst im Laufe der Differenzierung exprimieren die T-Zellen den T-Zell-Rezeptor für die Antigenerkennung sowie die charakteristischen CD4- oder CD8-Moleküle und den CD3-Komplex. Ähnlich wie die Gene für die beiden Antikörperketten des B-Zell-Rezeptors rearrangiert werden, entsteht in den unreifen T-Zellen ein Prä-T-Zell-Rezeptor, der sich zu einem Komplex mit CD3-Oberflächenmolekülen zusammenlagert. Dies ist wiederum das Signal für die Zelle, sich zu teilen und sowohl das CD4- als auch das CD8-Antigen zu exprimieren. Diese doppelt-positiven Zellen besitzen nur eine Lebenszeit von drei bis vier Tagen, es sei denn, ihr T-Zell-Rezeptor findet einen Bindungspartner in Form einer Epithelzelle im Cortex des Thymus, die ein MHC-Molekül (MHC: major histocompatibility complex) mit einem Peptid trägt. Durch das Interaktionssignal reifen die T-Zellen entweder zu einfachen CD4- oder CD8-positiven T-Zellen (Abb. 2).
Diesen Prozess bezeichnet man als positive Selektion oder auch MHC-Restriktion. Nur 10 bis 30% der T-Zellen erkennen mit ihren Rezeptoren ein Selbst-Peptid in einem MHC-Komplex, der auf Epithelzellen des Thymus-Cortex exprimiert wird. Ist die Bindung an eines der ubiquitär exprimierten Selbst-Antigene jedoch zu stark, werden die entsprechenden T-Zellen in die Apoptose geschickt.
Die bereits positiv selektierten, inzwischen nur noch entweder CD4- oder CD8-positiven T-Zellen wandern anschließend in die Medulla des Thymus (Abb. 2). Dort befinden sich ebenfalls Epithelzellen und außerdem dendritische Zellen, die beide verschiedenste Autoantigene des Körpers präsentieren. Mithilfe des Transkriptionsfaktors AIRE (Autoimmun-Regulator) wird erreicht, dass die verschiedenen antigenpräsentierenden Zellen auch Gene exprimieren, die eigentlich nur in ganz anderen Organen vorkommen. Dieser Effekt wird auch als promiskuitive Genexpression bezeichnet und dient dazu, die T-Zellen tolerant gegenüber organspezifisch exprimierte Antigene des Körpers zu machen.
Auch in der Medulla werden all jene T-Zellen, die mit ihrem Rezeptor von dendritischen Zellen oder Thymus-Epithelzellen exprimierte Antigene erkennen, in die Apoptose geschickt. Dies bezeichnet man als negative Selektion, da auf diese Weise die selbstreaktiven T-Zellen eliminiert werden. Am Ende der Positiv- und Negativselektion steht ein reifes T-Zell-Repertoire, das sowohl auf das MHC-Muster des individuellen Menschen als auch auf Selbsttoleranz abgestimmt ist und das nur noch aus ca. 1 bis 2% der ursprünglich angelegten Thymozyten besteht.
Ebenso wie bei den B-Zellen gelangen auch vereinzelt selbstreaktive T-Zellen in die Peripherie. Bei diesen Zellen kommen die peripheren Toleranzmechanismen Anergie und immunologische Ignoranz zum Tragen, die einen Angriff der körpereigenen Strukturen verhindern.
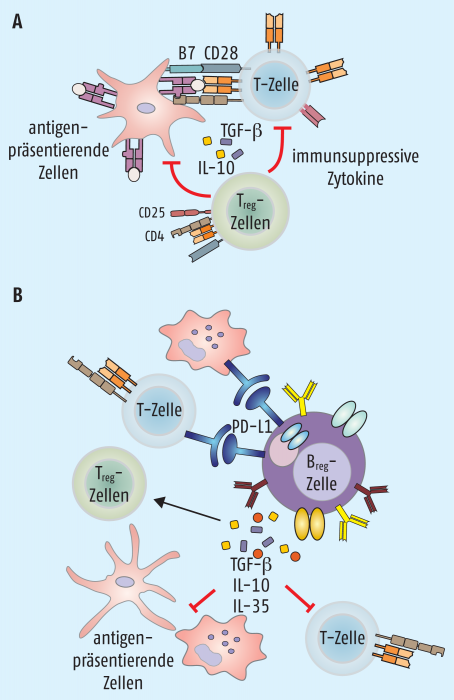
Zusätzlich kann die Differenzierung zu natürlichen regulatorischen T-Zellen (Treg-Zellen) autoreaktive T-Lymphozyten in der Peripherie inhibieren. Treg-Zellen können immer dann entstehen, wenn T-Zellen starke Signale empfangen, die jedoch gerade noch keine Apoptose im Sinne einer negativen Selektion induzieren. Diese Zellen sind charakterisiert durch die Produktion von immunsuppressiven Faktoren wie IL-10 oder TGF-β und unterdrücken eine TH1- und eine TH2-Antwort. Unter bestimmten Bedingungen können im Rahmen einer tolerogenen Immunantwort induzierte Treg-Zellen entstehen, die sich aus sogenannten Tr1- und TH3-Zellen zusammensetzen. Die Mechanismen, über die Treg-Zellen die Aktivität der Immunzellen regulieren, sind sehr unterschiedlich: Neben der Expression der antiinflammatorischen Zytokine TGF-β oder IL-10 kann über eine CTLA4-CD80-Interaktion die Expression costimulatorischer Proteine auf antigenpräsentierenden Zellen reduziert werden, was sich indirekt auf die Stimulation der T-Zellen auswirkt. Und schließlich exprimieren einige Treg-Zellen Granzym und Perforin und können direkt aktivierte T-Effektor-Zellen abtöten (Abb. 3 A).
Relativ neu ist die Entdeckung von regulatorischen B-Zellen, die ebenfalls in die Immunantwort eingreifen, indem sie die Differenzierung von TH1- und TH17-Zellen inhibieren und die Entwicklung von Treg-Zellen stimulieren. Außerdem können sie die Aktivierung von dendritischen Zellen und Makrophagen verhindern und T-Effektor-Zellen über direkten Zellkontakt in die Apoptose schicken (Abb. 3 B).
Da den B- und T-Lymphozyten in den zentralen Reifungsorganen (Knochenmark und Thymus) nicht alle denkbaren Autoantigene präsentiert werden, haben sich Mechanismen etabliert, die zur peripheren Toleranz führen. Erkennen die in die Peripherie ausgewanderten B- und T-Zellen erstmals ein Autoantigen, können verschiedene Wege eingeschlagen werden:
- Deletion (Apoptose) durch Induktion von activation-induced cell death (AICD), z. B. nach Restimulierung von kürzlich aktivierten CD4+-T-Zellen, oder wenn viele T-Zellen in einem bestimmten Milieu aktiviert werden (Deletion-induzierte Toleranz).
- Anergie mit der Folge, dass die Affinität des T-Zell-Rezeptors zum MHC/Peptid-Komplex zu schwach ist oder dass eine Interaktion mit CTLA-4 eingegangen wird.
- Immunologische Ignoranz gegenüber Antigenen, die in zu geringer Menge präsentiert werden oder die in immunprivilegierten Orten des Körpers (Auge, Gehirn, Hoden, Uterus) exprimiert werden.
- Suppression durch regulatorische T- und B-Zellen.
Autoimmunität
Eine Homöostase zwischen aktivierten Immunzellen (z. B. T- und B-Zellen) und regulatorischen T- und B-Zellen sorgt normalerweise für ein ausgeglichenes Verhältnis zwischen Toleranz gegenüber dem eigenen Gewebe und Immunantwort gegenüber Fremdstoffen und Pathogenen. Wichtiger Bestandteil einer geregelten Immunantwort gegen körperfremde Epitope sind selbstreaktive Immunzellen und Antikörper, die ein Überschießen der Immunantwort verhindern. Diese Art der Autoimmunität wird als physiologisch bezeichnet und ist selbstlimitierend.
Demgegenüber führt die länger vorliegende, chronische, pathologische Autoimmunität zu deutlichen Zerstörungen der betroffenen Gewebe. Je nachdem, wie weit verbreitet diese Gewebezerstörungen im Körper auftreten, lassen sich systemische und organspezifische Autoimmunerkrankungen unterscheiden (Tab. 1).
Erkrankung |
betroffenes Organ/Zielstruktur |
|---|---|
organspezifische Autoimmunerkrankungen | |
multiple Sklerose |
Myelinscheide der Nervenfasern |
Diabetes mellitus Typ 1 |
Inselzellen der Bauchspeicheldrüse |
Colitis ulcerosa |
Darmschleimhaut |
Pemphigus vulgaris |
oberste Hautschicht; Epidermis |
Myasthenia gravis |
Acetylcholin-Rezeptoren an motorischen Endplatten |
Morbus Basedow |
TSH-Rezeptoren der Schilddrüse |
systemische Autoimmunerkrankungen (Erkrankungen des entzündlich-rheumatischen Formenkreises) | |
rheumatoide Arthritis |
chronische Polyarthritis |
Lupus erythematodes (SLE) |
Reaktionen gegen zahlreiche Organe |
Polymyositis |
Entzündung der Muskulatur |
Sjögren-Syndrom |
exokrine Drüsen |
Sklerodermie |
Bindegewebsverhärtung von Haut, Gefäßen und inneren Organen |
systemische Vaskulitiden |
Entzündung der Gefäße |
Anti-Phospholipid-Syndrom |
Störung in der Blutgerinnung |
Manche organspezifischen Autoimmunerkrankungen können sogar gleichzeitig in einem Patienten auftreten: z. B. ist ein systemischer Lupus erythematodes gemeinsam mit dem Sjögren-Syndrom, oder eine Thyreoid-Autoimmunerkrankung mit Vitiligo gefunden worden.
Der Verlauf einer Autoimmunreaktion lässt sich in drei Phasen einteilen: Durch genetische, umweltbedingte und/oder immunologische bzw. mikrobielle Faktoren kommt es zunächst zur Initiation. In der Propagation reagiert das Immunsystem auf körpereigene Antigene und durch Effekte wie Epitop-Spreading, eine gesteigerte Zytokin-Produktion sowie ein Ungleichgewicht zwischen T-Effektor- und T-Regulator-Zellen wird diese Reaktion über einige Zeit aufrecht gehalten. Idealerweise steuert der Körper selbst wieder gegen und bringt im Falle der physiologischen Autoimmunität während der Resolution die Verschiebung der Immunreaktion durch inhibitorische Signalwege oder durch Treg- bzw. Breg-Zellen wieder in Ordnung.
Initiation
Genetische Prädisposition.
Circa 30% der genetischen Prädisposition ist allein auf die Varianz in der HLA-Region zurückzuführen. Über 400 Gene befinden sich innerhalb des ungefähr 7,2 MBp großen genomischen Segments, die nicht nur an der Prozessierung und Präsentation von Antigenfragmenten beteiligt sind, sondern die auch Zellaktivierung, Entzündungsreaktion und andere zentrale Effekte der angeborenen und adaptiven Immunantwort steuern. Ganz prominent sind natürlich die MHC-Proteine selbst betroffen (Tab. 2). Nachdem diese Proteinkomplexe den Immunzellen Peptid-Antigene präsentieren, lässt sich leicht vorstellen, dass sie dabei ganz bestimmte Aminosäureabfolgen präferieren. Bei manchen MHC-Molekülen können dies wiederum Teile körpereigener Strukturen sein – der erste Schritt hin zu einer Autoimmunerkrankung.
Erkrankung |
HLA-Allel |
Autoantigen |
|---|---|---|
Lupus erythematodes (SLE) |
DR2, DR3 |
ds/ssDNA |
Diabetes mellitus Typ 1 |
DR3, DR4 |
Insulin, Glutamat-Decarboxylase Isoform 65 |
Graves-Erkrankung |
DR3 |
TSH-Rezeptor |
rheumatoide Arthritis |
DR1, DR4 |
Kollagen Typ II |
Eine andere, recht sicher identifizierte genetische Korrelation kann über den Transkriptionsfaktor AIRE (Autoimmun-Regulator) hergestellt werden. Ist dieses Protein verändert und nicht mehr funktionsfähig, können im Thymus die gewebespezifischen Antigene nicht mehr effizient promiskuitiv exprimiert werden. Dadurch fehlt den T-Lymphozyten ein entscheidender Lernprozess für die körpereigenen Proteine, und autoreaktive Immunzellen werden nicht mehr korrekt aussortiert.
Durch genomweite Assoziationsstudien (GWAS) sind inzwischen noch zahlreiche andere Gene als potenzielle Korrelate mit Autoimmunerkrankungen gefunden worden. Alle Proteine, die in irgendeiner Form an der Entwicklung und Aktivierung der Immunzellen beteiligt sind, können wahrscheinlich in mutierter Form eine Autoimmunerkrankung fördern.
Eine Art der genetischen Ausstattung – sogar ganz ohne Mutationen – ist ebenfalls mit einem erhöhten Risiko für Autoimmunerkrankungen assoziiert: Frauen sind wesentlich häufiger z. B. von systemischem Lupus erythematodes, Sjögren-Syndrom, Sklerodermie oder Morbus Basedow betroffen als Männer.
Umweltfaktoren. Üblicherweise reicht eine genetische Prädisposition noch nicht aus, um tatsächlich eine Autoimmunerkrankung auszulösen. Unterstützt werden kann der Prozess z. B. über verschiedene Umweltfaktoren. Bei physikalischen und chemischen Umweltfaktoren wie UV-Strahlung oder bestimmten Lösungsmitteln ist es nochmal ungleich schwerer, einen kausalen Zusammenhang zum Auftreten bestimmter Autoimmunerkrankungen herzustellen. Erwiesen ist, dass Zigarettenrauch an der Entstehung von systemischem Lupus erythematodes und rheumatoider Arthritis beteiligt sein kann. Die Vermutung liegt nahe, dass die Entstehung anderer Autoimmunerkrankungen ebenfalls durch Rauchen unterstützt wird. Halbwegs plausibel scheint der Zusammenhang zwischen UV-Strahlung und systemischem Lupus erythematodes: Da UV-B-Strahlung in Hautzellen eine Apoptose induziert, kann es zur Freisetzung von Autoantigenen und proinflammatorischen Zytokinen kommen, die dann wiederum eine Autoimmunerkrankung triggern.
Interessante Beobachtungen wurden auch mit Alkohol im Zusammenhang mit Autoimmunerkrankungen gemacht. Hier deuten verschiedene Studien darauf hin, dass leichter bis moderater Konsum sogar vor rheumatoider Arthritis und systemischem Lupus erythematodes (SLE) schützen könnte. Demgegenüber gibt es Verdachtsmomente, dass eine Exposition mit organischen und anorganischen Lösungsmitteln das Auftreten eines systemischen Lupus erythematodes oder einer rheumatoiden Arthritis fördert.
Arzneistoffe.
Die ersten Hinweise, dass bestimmte Arzneistoffe zu Autoimmunerkrankungen führen können, stammen von Beobachtungen aus dem Jahr 1945 an Patienten, die Sulfadiazin einnahmen und SLE-ähnliche Symptome zeigten. Und auch mit Hydralazin konnten wenige Jahre später ähnliche Folgen gezeigt werden. Mittlerweile nimmt man von mehr als 100 Wirkstoffen, die zu sehr unterschiedlichen Arzneistoff-Kategorien gehören, an, dass sie mit dem Auftreten von Autoimmunerkrankungen assoziiert sind. Diese sogenannte drug-induced autoimmunity (DIA, Arzneimittel-induzierte Autoimmunreaktion) ist üblicherweise eine Typ-B-Wirkstoffreaktion, die unvorhergesehen bei einer Therapie auftritt und durch etliche, weitere Faktoren, wie z. B. auch eine genetische Prädisposition, beeinflusst wird. Eine Arzneimittel-induzierte Autoimmunreaktion hört typischerweise nach Absetzen des Arzneimittels wieder auf. Die häufigste DIA ist systemischer Lupus erythematodes, aber auch rheumatoide Arthritis, Polymyositis, Myasthenia gravis, autoimmune Hepatitis, autoimmune Thyreoiditis und etliche andere können durch Arzneistoffe induziert werden (Tab. 3).
Wirkstoffkategorie |
Arzneistoff |
Autoimmunerkrankung |
|---|---|---|
Antihistaminika |
Cimetidin |
autoimmune hämolytische Anämie |
antiinflammatorische Wirkstoffe |
Ibuprofen |
autoimmune hämolytische Anämie |
Mesalazin |
SLE, idiosynkratische Thrombozytopenie, autoimmune Hepatitis |
|
Sulfasalazin |
SLE, Vaskulitis |
|
Biologika |
Adalimumab |
SLE, Vaskulitis, Antiphospholipid-Syndrom |
Etanercept |
SLE, Vaskulitis, Sarcoidose, granulomatöse Lungenkrankheit |
|
Golimumab |
subakut kutaner Lupus erythematodes (SCLE) |
|
Infliximab |
SLE, Vaskulitis, interstitielle Lungenkrankheit, entzündliche Myopathie |
|
Interferon α |
Thyroid-Autoimmunität, SLE, Vaskulitis, autoimmune Hepatitis |
|
Interferon β |
Thyroid-Autoimmunität, SLE, Vaskulitis |
|
Interleukin 2 |
Thyroid-Autoimmunität, chronisch-entzündliche Arthritis |
|
Antibiotika |
Cefuroxim |
Pemphigus erythematosus, SLE |
Isoniazid |
SLE, autoimmune hämolytische Anämie |
|
Nalidixinsäure |
SLE, autoimmune hämolytische Anämie |
|
Penicillin |
autoimmune hämolytische Anämie |
|
Streptomycin |
SLE, autoimmune hämolytische Anämie |
|
Tetracyclin |
SLE, Vaskulitis, autoimmune hämolytische Anämie |
|
Antiarrhythmika |
Acecainid |
SLE |
Procainamid |
SLE |
|
Propafenon |
SLE |
|
Antihypertonika |
Acebutolol |
SLE |
Atenolol |
SLE |
|
Captopril |
SLE, autoimmune Thrombozytopenie |
|
Enalapril |
SLE, Vaskulitis |
|
Hydralazin |
SLE, Vaskulitis |
|
Metoprolol |
SLE |
|
Propranolol |
SLE |
|
Statine |
Atorvastatin |
SLE, Dermatomyositis, Polymyositis |
Fluvastatin |
SLE, Dermatomyositis, Polymyositis |
|
Lovastatin |
SLE, Dermatomyositis |
|
Pravastatin |
SLE, Dermatomyositis, Polymyositis |
|
Simvastatin |
SLE, Dermatomyositis, Polymyositis, Lichen planus pemphigoides |
|
Antikonvulsiva |
Carbamazepin |
SLE |
Phenytoin |
SLE, lineare IgA bullöse Erkrankung |
|
Antiparkinsonmittel |
Levodopa |
autoimmune hämolytische Anämie, SLE, Thrombozytopenie |
SLE: systemischer Lupus erythematodes | ||
Darm-Mikrobiom. Viel wird in letzter Zeit über das Mikrobiom des Menschen und seine Beteiligung an verschiedenen Krankheitsgeschehen geforscht und beschrieben. An vorderster Front sind die Bakterien in unserem Darm auch an der Reifung des angeborenen Immunsystems beteiligt. Außerdem steuern sie die Entwicklung von z. B. TH17- oder Treg-Zellen. In Mäusen führt die Verabreichung der sogenannten „veränderten Schaedler Flora“, also einem Cocktail aus acht unterschiedlichen Bakterienstämmen zu einer erhöhten Zahl an Treg-Zellen im Gastrointestinaltrakt der Mäuse.
Vergleicht man das Mikrobiom gesunder Menschen mit dem von Patienten, die unter einer Autoimmunerkrankung leiden, lassen sich Veränderungen in der Zusammensetzung der Bakterienpopulation feststellen. Noch ist es allerdings zu früh, um hier wirklich auch von Kausalitäten zu sprechen. Jedoch zielen einige Therapieansätze bei Autoimmunerkrankungen in Richtung Mikrobiom. Beispielsweise gelten Doxycyclin oder Minocyclin als recht wirksame DMARDs bei der Behandlung von Rheumapatienten im frühen Krankheitsstadium. Und Ciprofloxacin wird häufig zur Behandlung des systemischen Lupus erythematodes eingesetzt. Man könnte die Anwendung dieser Antibiotika auch als Regulierung der Zusammensetzung der Darmflora interpretieren. In die andere Richtung, nämlich über die Anwendung von Probiotika, lässt sich ebenfalls eine Therapie vorstellen. Verabreicht man Bakterien der Clostridium-coccoides-Gruppe, Faecalibacterium prausnitzii oder Bifidobacterium, lassen sich Treg-Zellen anregen und die Symptome einer Darmerkrankung verbessern – zumindest im Tiermodell. Erste klinische Studien deuten auch auf eine Wirksamkeit bei Patienten hin, wenn Lactobacillus casei bei rheumatoider Arthritis oder ein Bakteriencocktail aus drei Bifidobacterium- und vier Lactobacillus-Stämmen sowie Streptococcus salivarius sp. thermophilus angewendet wurden.
Natürlich beeinflusst die Ernährung ebenfalls das Darm-Mikrobiom. Klinische Studien deuten darauf hin, dass vegetarische Ernährung und Fasten Krankheiten lindern können, während westliche Ernährung, hier vor allem fettreiche Lebensmittel, verschiedene Krankheiten verschlimmert.
Infektionen. Was bereits seit Langem vermutet wird und für einige Autoimmunerkrankungen bereits klar gezeigt werden konnte, ist die Verknüpfung von bestimmten Infektionen mit einer Reaktivität des Immunsystems gegen Selbstantigene. Diese Assoziation betrifft vor allem bakterielle und virale Infektionen, aber auch Mykosen scheinen problematisch zu sein. Zumindest konnte eine Reaktivität gegenüber Candida albicans gezeigt werden. Die Mechanismen, über die die Pathogene die Autoimmunerkrankung stimulieren, sind vielfältig:
- molekulare Mimikry,
- Epitop-Spreading,
- Stimulation von Mustererkennungsrezeptoren und Aktivierung von weiteren Immunzellen (Bystander-Aktivierung),
- virale Persistenz und polyklonale Aktivierung von B-Zellen sowie
- autoinflammatorische Aktivierung des angeborenen Immunsystems.
Molekulare Mimikry
bedeutet, dass Epitope von Pathogenen sehr ähnliche Strukturen aufweisen wie bestimmte Autoantigene. Nach der Infektion werden dem Immunsystem Peptide des Virus/Bakteriums präsentiert – mit einer gewissen Wahrscheinlichkeit auch dasjenige mit einer großen Homologie zum Selbstantigen (Tab. 4). Sind dann Antikörper oder T-Zellen gegen das Epitop mobilisiert, können sie gegebenenfalls auch an das Peptid des Autoantigens binden und eine Immunreaktion induzieren (Abb. 4).
Krankheitserreger und menschliche Antigene |
identische Peptidsequenz |
|---|---|
|
menschliches Zytomegalie-Virus IE2
HLA-DR-Moleküle
|
PDPLGRPDED
VTELGRPDAE
|
|
Poliovirus VP2
Acetylcholin-Rezeptor
|
STTKESRGTT
TVIKESRGTK
|
|
Papillomavirus E2
Insulin-Rezeptor
|
SLHLESLKDS
VYGLESLKDL
|
|
Klebsiella pneumoniae Nitrogenase
HLA-B27-Moleküle
|
SRQTDREDE
KAQTDREDL
|
|
Adenovirus 12 E1B
Alfa-Gliadin
|
LRRGMFRPSQCN
LGQGSFRPSQQN
|
|
HIV p24
menschliches IgG
|
GVETTTPS
GVETTTPS
|
|
Masernvirus P3
Myelinprotein
|
EISDNLGQE
EISFKLGQE
|
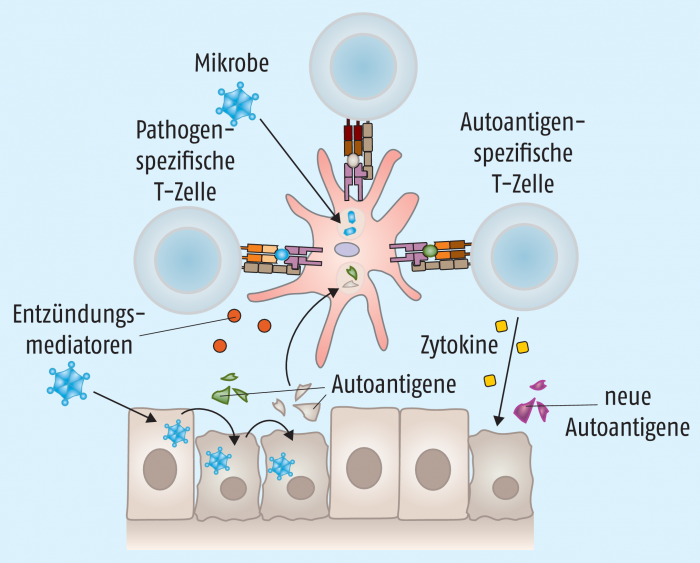
Beim Epitop-Spreading erweitert sich das Spektrum der erkannten Selbstantigene (Abb. 5). Kommt es bei einer bakteriellen oder viralen Infektion zur Zerstörung von Körperzellen, können bis dahin versteckte körpereigene Antigene freigesetzt werden. Antigenpräsentierende Zellen können diese Antigene wiederum aufnehmen, prozessieren und T-Zellen präsentieren, wodurch die Autoimmunreaktion induziert wird.
Stimuliert ein Pathogen bei einer Infektion Toll-like-Rezeptoren oder andere Pattern-Recognition-Rezeptoren auf antigenpräsentierenden Zellen, kommt es zur Produktion proinflammatorischer Zytokine und dadurch wiederum zu Gewebeschäden. Lokal sind nun nicht nur Antigene des Pathogens, sondern auch freigesetzte Gewebeantigene präsent, die jeweils spezifische T-Zellen aktivieren können – auch autoreaktive T-Zellen, die an der ursprünglichen Infektion gar nicht beteiligt waren: sogenannte Bystander oder „Zuschauer“ (Abb. 6). Verstärkt wird dieser Prozess durch Super-Antigene, die von einigen Viren, wie Epstein-Barr-Virus oder Retroviren, und vor allem von Bakterien wie Staphylokokken, Streptokokken oder Mycoplasmen gebildet werden können. Diese Superantigene sind in der Lage, sowohl an MHC-II-Moleküle auf antigenpräsentierenden Zellen als auch an T-Zell-Rezeptoren (TCR) auf T-Zellen zu binden. Dadurch kommt es unabhängig von der Peptidbeladung des MHC-II zu einer Aktivierung der T-Zelle – eventuell auch einer unbeteiligten autoreaktiven Bystander-T-Zelle (Abb. 6).
Manche Viren, wie das Epstein-Barr-Virus, sind in der Lage, für längere Zeit in B-Zellen zu persistieren und dadurch diese Immunzellen zu aktivieren und zur Proliferation anzuregen. Im Zuge dieser Aktivierung und Proliferation kommt es durch die somatische Hypermutation innerhalb der B-Zellen zu einer gewissen Varianz der produzierten Antikörper, die sich eventuell auch gegen Autoantigene richten können – ein Prozess, den man als polyklonale Aktivierung der B-Zellen bezeichnen kann.
Während einer normalen Immunantwort auf eine Infektion kommt es immer zur Kommunikation zwischen angeborenem und adaptivem Immunsystem. Entgleisungen des adaptiven Teils sind die bereits erwähnten verschiedenen Autoimmunerkrankungen. Man kennt jedoch auch entsprechende Entgleisungen des angeborenen Immunsystems. Diese sogenannten autoinflammatorischen Erkrankungen werden als periodisch auftretende Fiebersyndrome bezeichnet, wie z. B. das familiäre mediterrane Fieber oder das Cryopyrin-assoziierte periodische Syndrom. Bei diesen Erkrankungen geht man davon aus, dass das Inflammasom, das nach einer ersten Infektion unspezifisch für das Anschalten der Entzündungsantwort in den Immunzellen sorgt, dysreguliert ist. Mittlerweile sind Hinweise vorhanden, die ein dysreguliertes Inflammasom auch mit Autoimmunerkrankungen in Verbindung bringen.
Entzündungsreaktionen werden oftmals im Zuge von aktiven Immunisierungen mithilfe der Adjuvanzien induziert. Kein Wunder also, dass manche Adjuvanzien (und einige Impfstoffe) in Verdacht stehen, autoinflammatorische Erkrankungen zu provozieren und darüber letztlich sogar Autoimmunerkrankungen. Allerdings ist die Anzahl der Fälle von Autoimmunerkrankungen, die irgendwie mit Impfungen in Verbindung gebracht werden, sehr gering, und die Vorteile einer Impfung überwiegen bei Weitem das Risiko einer Autoimmunerkrankung.
Eine andere Ursache für die autoinflammatorischen Erkrankungen kann man hingegen wahrscheinlich leichter meiden: Silikon-Implantate. Vor allem diejenigen, die undicht sind, erhöhen bei einer entsprechenden genetischen Prädisposition erheblich das Risiko.
Propagation
Mit den Infektionen durch verschiedene Pathogene haben wir bereits die Grenze von Initiation zur Propagation der Autoimmunerkrankung überschritten. Ohne die Reaktion des Immunsystems auf die Infektion käme es nicht zur Zytokin-Produktion, nicht zur Aktivierung der T- und B-Zellen und nicht zu Immunzellen, die sich dann auch gegen Autoantigene richten. Unglücklicherweise können diese Autoantigene nicht entfernt werden – als körpereigene Moleküle sind sie allgegenwärtig. Im Gegenteil: Ist erst einmal die Immunantwort entfacht, kommt es durch Epitop-Spreading noch zu einer Erweiterung des Autoantigenrepertoires – ein Circulus vitiosus, der nicht mehr so leicht zu durchbrechen ist. Finden sich dann zudem nicht genügend regulatorische Mechanismen wie antiidiotypische Antikörper, die die Autoantikörper in ihrer Reaktivität einschränken, oder Breg- bzw. Treg-Zellen, die die autoreaktiven Immunzellen inhibieren, kommt es zur massiven Schädigung der eigenen Organe.
Therapie
Autoimmunerkrankungen lassen sich nicht wirklich kausal therapieren – zu viele Mitspieler sind daran beteiligt. Der Wirkstoff TGN1412, der 2006 als CD28-Superagonist in die Geschichte einging, war ursprünglich entwickelt worden, um die Treg-Zellen zu stimulieren und darüber Autoimmunerkrankungen zu heilen. In der Phase-I-Studie führte dieser Antikörper zu einer massiven Zytokin-Ausschüttung im Körper der Probanden. Aber immerhin zeigen diejenigen Substanzen, die recht gezielt an verschiedenen Rädchen innerhalb der Immunreaktion angreifen, gewisse Wirkung. Besonders hervorzuheben sind hier die Biologika: So wirken TNF-α-Inhibitoren sehr gut bei rheumatoider Arthritis und Psoriasis-Arthritis, oder Ustekinumab inhibiert sowohl das Signal von IL-12 als auch von IL-23 und wird unter anderem gegen Plaque-Psoriasis angewendet (Tab. 5). Mit der genaueren Kenntnis der an der Autoimmunantwort beteiligten Moleküle und Zellen hat sich das Spektrum an gezielt einsetzbaren Wirkstoffen erheblich erweitert. So kann z. B. der Antikörper gegen CD20 auf B-Zellen nicht nur bei B-Zell-Tumoren eingesetzt werden, sondern auch bei Autoimmunerkrankungen wie rheumatoider Arthritis und multipler Sklerose. Damit kommt man weg von den zytostatisch auf Immunzellen wirkenden Arzneistoffen wie Cyclophosphamid, Methotrexat oder Azathioprin.
Wirkstoff |
Molekül |
Zielstruktur |
|---|---|---|
Abatacept |
Fusionsprotein aus CTLA-4 und Antikörper-Fc-Teil |
CD80 und CD86 |
Adalimumab |
humaner Antikörper |
TNF-α |
Alemtuzumab |
humanisierter Antikörper |
CD52 auf B- und T-Zellen |
Anakinra |
Interleukin-1-Rezeptorantagonist |
IL-1 |
Basiliximab |
chimärer Antikörper |
CD25 auf aktivierten T-Zellen |
Belimumab |
humaner Antikörper |
BLys |
Certolizumab pegol |
pegyliertes, humanisiertes Fab-Fragment |
TNF-α |
Daclizumab |
humanisierter Antikörper |
CD25 auf aktivierten T-Zellen |
Etanercept |
Fusionsprotein aus TNF-R und Antikörper-Fc-Teil |
TNF-α |
Golimumab |
humaner Antikörper |
TNF-α |
Infliximab |
chimärer Antikörper |
TNF-α |
Ixekizumab |
humanisierter Antikörper |
IL-17A |
Natalizumab |
humanisierter Antikörper |
α4-Integrin |
Peginterferon beta 1a |
pegyliertes Interferon β |
|
Rituximab |
chimärer Antikörper |
CD20 auf B-Zellen |
Secukinumab |
humaner Antikörper |
IL-17A |
Tocilizumab |
humanisierter Antikörper |
IL-6-Rezeptor |
Ustekinumab |
humaner Antikörper |
IL-12, IL-23 |
Vedolizumab |
humanisierter Antikörper |
α4β7-Integrin |
Aber: Ein therapeutischer Eingriff in das Immunsystem sollte immer auch kritisch beobachtet werden, nicht erst seit dem Vorfall mit TGN1412. Wenn derzeit so viele Hoffnungen in die neuen Checkpoint-Inhibitoren für die Krebstherapie gelegt werden, sollte nicht vergessen werden, dass es sich hier um eine Stimulation der T-Zellen handelt, die sich im Idealfall gegen die Tumorzellen richten, im schlechteren Fall gegen Autoantigene. Insofern sollte man vor allem etwas vorsichtiger sein mit einem dauerhaften genetischen Knockout von PD-1 in Blutzellen, wie er momentan in einer klinischen Studie in China getestet wird. |
Autoren
Prof. Dr. Theo Dingermann ist Seniorprofessor am Institut für Pharmazeutische Biologie an der Goethe-Universität Frankfurt.
Dr. Ilse Zündorf ist dort als akademische Oberrätin tätig.
Institut für Pharmazeutische Biologie Biozentrum, Max-von-Laue-Straße 9 60438 Frankfurt/Main
Zum Weiterlesen
Immunologie – Grundlagen und Wirkstoffe
von Angelika Vollmar, Ilse Zündorf und Theodor Dingermann
2., völlig neu bearbeitete und erweiterte Auflage 2012.
Wissenschaftliche Verlagsgesellschaft Stuttgart
Zu beziehen über:
Deutscher Apotheker Verlag, Birkenwaldstraße 44, 70191 Stuttgart
Telefon 0711 2582-341, Telefax 0711 2582-290,
E-Mail: service@deutscher-apotheker-verlag.de



1 Kommentar
Autoimmunkrankheiten durch Implantate bzw. Zahnersatz.
von Menschenrechtler am 02.01.2020 um 23:22 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Das Kommentieren ist aktuell nicht möglich.