













- DAZ.online
- DAZ / AZ
- DAZ 30/2016
- Kapseln herstellen

Foto: kasezo – 123rf.com
Rezeptur
Kapseln herstellen
Unterschiedliche Methoden – unterschiedliche Herausforderungen
Dieser Artikel beinhaltet vor allem theoretische Betrachtungen und Erklärungen zu den verschiedenen Methoden zur Kapselfüllung – von Pulvern bis Schmelzen. Ein weiterer Artikel in dieser Ausgabe der DAZ („Die Kapsel – Eine einfache Arzneiform?“) hat als Schwerpunkt die Befüllung von Kapseln mit „klassischen“ Pulvermischungen und beinhaltet vor allem experimentelle Ergebnisse.
Verschiedene Arten von Kapseln
Das Europäische Arzneibuch kennt Hartkapseln, Weichkapseln, magensaftresistente Kapseln, Kapseln mit veränderter Wirkstofffreisetzung und Oblatenkapseln. Für die Rezeptur relevant sind hierbei vor allem die Hartkapseln. Sie bestehen in den meisten Fällen aus Gelatine, es gibt allerdings auch Varianten aus Cellulose oder Hypromellose. Zur Füllung dienen meist Pulver, bei denen der Wirkstoff vor allem mit Mannitol, aber auch mit Lactose-Monohydrat, mikrokristalliner Cellulose, Glucose oder Stärke (eventuell unter Zusatz eines Fließregulierungsmittels wie hochdispersem Siliciumdioxid) vermischt wird. Wesentliche Informationen zu den unterschiedlichen Hüllmaterialien und Füllstoffen können dem DAC/NRF entnommen werden. Sie werden in diesem Artikel nicht weiter thematisiert.
Gesicherte und reproduzierbare Qualität
Anforderungen an Kapseln
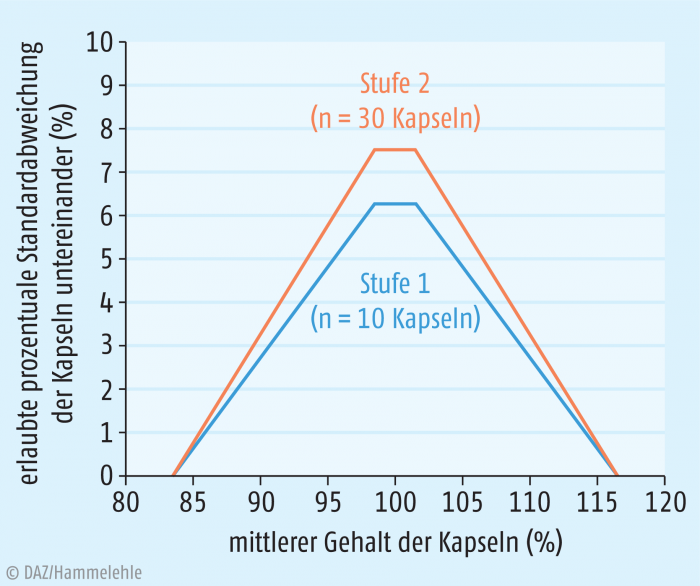
Die wichtigste Anforderung an Kapseln lässt sich in einfachen Worten ausdrücken: In jeder Kapsel soll die verordnete Wirkstoffmenge enthalten sein. In Bezug auf die Prüfmethoden des Europäischen Arzneibuchs bedeutet dies für industriell hergestellte Kapseln, dass die Prüfung „2.9.40 Gleichförmigkeit einzeldosierter Arzneiformen“ bestanden werden muss. Hierbei wird je nach Einzeldosis (Grenze bei 25 mg) und Wirkstoffanteil (Grenze bei 25%) unterschieden, ob zur Durchführung der Prüfung in erster Linie der Gehalt geprüft wird, oder ob eine Prüfung anhand eines mittleren Gehaltes und Einzelmassen möglich wird. Wichtig ist jedoch, dass hier immer eine Gehaltsbestimmung erfolgt: entweder an einer repräsentativen Stichprobe der Charge oder an zehn (bzw. 30) Einzelkapseln. Das weitere Vorgehen wird im Detail im Arzneibuch beschrieben. Die Prüfung wird auf zwei Stufen bewertet: Für die erste Stufe werden zehn Kapseln geprüft. Wird die erste Stufe nicht bestanden, so werden 20 weitere Kapseln geprüft. Als Anforderung gilt letztlich, dass die Kapseln einen umso gleichmäßigeren Gehalt bzw. eine umso gleichmäßigere Masse aufweisen müssen, je weiter sich der mittlere Gehalt vom Sollgehalt entfernt. Liegt der mittlere Gehalt außerhalb des Bereiches von 83,5% bis 116,5%, kann die Prüfung auf Stufe 1 nicht mehr bestanden werden. Auf Stufe 2 gelten prinzipiell ähnliche Anforderungen, die maximal zulässige Standardabweichung steigt jedoch etwas an (siehe Abbildung 1). Zusätzlich muss nun der Gehalt der einzelnen Kapsel beachtet werden: Keine Kapsel darf einen Einzelgehalt von unter 73,875% oder über 126,875% aufweisen.
Im Rezepturmaßstab wird die Gehaltsbestimmung nicht gefordert, da es sich um eine zugelassene Ausnahme handelt und eine Prüfung auch weiterhin nach der älteren Monografie 2.9.5 erfolgen kann (bei weniger als 2 mg oder 2% Wirkstoff nach 2.9.6, dies ist jedoch in der Rezeptur meist nicht der Fall). Hierzu wird der Inhalt von 20 Kapseln ausgewogen. Maximal zwei der einzelnen Massen dürfen um einen höheren als den angegebenen Prozentsatz von der mittleren Masse abweichen, jedoch keine mehr als das Doppelte. Beträgt die Masse des Kapselinhaltes unter 300 mg (z. B. zu erwarten bei der Füllung von Kapseln der Größe 1 mit Mannitol 35), liegt diese Grenze bei 10%. Bei einer Füllmasse von über 300 mg (z. B. zu erwarten bei der Füllung von Kapseln der Größe 0 mit Mannitol 35), liegt die Grenze bei 7,5%.
Weitere Anforderungen (Zerfallszeit, mikrobielle Belastung) können im Rezepturmaßstab im Allgemeinen als vergleichsweise eher unkritisch angesehen werden.
Bedauerlicherweise enthält der in DAZ Nr. 30 abgedruckte Text einen Fehler. In der Print-Ausgabe von DAZ Nr. 33 finden Sie dazu auf der S. 64 nachfolgende Korrektur:
"Erratum
In dem Beitrag 'Kapseln herstellen' in DAZ 2016, Nr. 30, S. 42, wurden bei den Angaben zur Prüfung auf Gleichförmigkeit der Masse (Ph.Eur. 2.9.5) auf Seite 43 die mögliche Abweichung von der Durchschnittsmasse (%) vertauscht. Richtig muss es heißen:
'... Beträgt die Masse des Kapselinhaltes unter 300 mg (z. B. zu erwarten bei der Füllung von Kapseln der Größe 1 mit Mannitol 35), liegt diese Grenze bei 10%.
Bei einer Füllmasse von über 300 mg (z. B. zu erwarten bei der Füllung von Kapseln der Größe 0 mit Mannitol 35), liegt die Grenze bei 7,5%.'
Wir bitten, den Fehler zu entschuldigen und bedanken uns bei unseren aufmerksamen Lesern für den Hinweis!"
Wir haben den Fehler hier im Text korrigert, da wir nicht möchten, dass die Information falsch verbreitet wird.
Prüfung von Kapseln in der Apotheke
Zur Prüfung von Rezepturarzneimitteln besagt die ApBetrO in § 7 (2): „Bei einem Rezepturarzneimittel kann von einer analytischen Prüfung abgesehen werden, sofern die Qualität des Arzneimittels durch das Herstellungsverfahren, die organoleptische Prüfung des fertig hergestellten Arzneimittels und, soweit vorgesehen, durch die Ergebnisse der Inprozesskontrollen gewährleistet ist.“ Die einzige praktikable Möglichkeit zur Prüfung von Kapseln in der öffentlichen Apotheke ist das Wiegen der komplett gefüllten Kapseln. Damit bleibt die eigentlich viel wichtigere Frage nach dem Wirkstoffgehalt (sowohl in Bezug auf Richtigkeit als auch auf die gleichmäßige Verteilung) in der Apotheke meist unbeantwortet. Dies macht es umso wichtiger, sich dieser Verantwortung bei der Herstellung von Kapseln bewusst zu sein.
Da die Gehaltsbestimmung der Kapseln nicht praktikabel ist (fehlende bzw. zu teure apparative Ausrüstung, vernichtende Prüfung), schlägt das NRF in den standardisierten Rezepturen folgende Vorgehensweisen zur Prüfung vor, die sich im Detail für die Rezepturen leicht unterscheiden:
Existiert ein Sollwert der Masse der Kapselfüllung (Befüllung mit Schmelzen), wird dieser mit der tatsächlichen mittleren Füllmasse verglichen. Eine Abweichung von 5% ist hierbei zu tolerieren. Zusätzlich wird eine vorgegebene Kapselanzahl (zehn Stück oder 20% der Charge, je nach Rezeptur) einzeln ausgewogen. Hierbei darf keine Einzelmasse zu weit (je nach Rezeptur 15% bis 20%) vom Sollwert (wenn vorhanden) oder Durchschnittswert aller Kapseln abweichen. Ist dies doch der Fall, werden alle restlichen Kapseln ebenfalls einzeln gewogen. Es gelten erweiterte Grenzen (etwa 20% bis 25%), die nun von allen Kapseln einzuhalten sind.
Die Allgemeinen Hinweise (DAC I.9.) beinhalten diesbezüglich nur die Empfehlung, dass die relative Standardabweichung der geprüften Kapselmassen einen Wert von 5% nicht überschreiten soll. Wie oben bereits erwähnt, ist dies aber nur ein Hinweis auf die gleichmäßige Befüllung der Kapseln – nicht jedoch auf die Homogenität der Mischung.
Methoden zur Kapselfüllung
Das NRF beschreibt unterschiedliche standardisierte Methoden zur Kapselfüllung. Es handelt sich dabei um Dronabinol-Kapseln nach NRF 22.7., Hydrochlorothiazid-Kapseln nach NRF 26.3., Thalidomid-Kapseln nach NRF 32.2. oder eben die klassischen Methoden zur Pulverfüllung nach Methode A bzw. Methode B aus Anlage G des DAC/NRF, wie sie auch in NRF 22.3. (3,4-Diaminopyridin-Kapseln) und NRF 21.5. (Neomycinsulfat-Kapseln) angewendet werden.
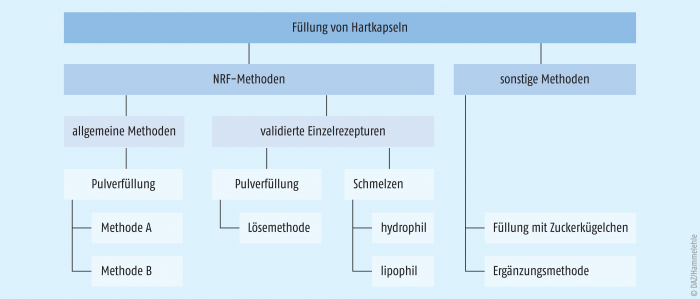
Eine Methode zur Herstellung von niedrig dosierten Hydrocortisonacetat-Kapseln befindet sich zudem derzeit in der Ausarbeitung. Der Zwischenstand kann dem (bei Bedarf ständig aktualisierten) Rezepturhinweis zu Hydrocortisonacetat in pädiatrischer Dosierung entnommen werden. Weiterhin wird die Möglichkeit zur Füllung mit Zuckerkügelchen erwähnt. Zusätzlich findet man in der Literatur eine Ergänzungsmethode beschrieben. Diese Methoden sind in Abbildung 2 nach dem Stand ihrer Validierung aufgeführt und werden im Folgenden näher beleuchtet.
NRF-standardisierte Methoden – Füllung mit Pulvermischungen
Grundprinzip
Das Prinzip der beiden beschriebenen Methoden unterscheidet sich zwar im Detail, ist aber grundlegend gleich: Das benötigte Pulvervolumen (Kalibriervolumen) wird mit Füllmittel experimentell ermittelt. Die benötigte Wirkstoffmenge wird mit Füllmittel so vermischt, dass das verwendete Volumen dem Kalibriervolumen entspricht. Hierzu sind verschiedene Schritte des Abmessens im Messzylinder und geeigneten Vermischens nötig. Schließlich wird die Pulvermischung gleichmäßig in die Leerkapseln gefüllt, und diese werden verschlossen.
Füllmittel
Das Standard-Füllmittel für Kapseln wird in der NRF-Stammrezeptur S.38. beschrieben. Es handelt sich um eine Mischung aus 99,5% Mannitol 35 und 0,5% hochdispersem Siliciumdioxid. Die Schüttdichte des Füllmittels wird (geprüft nach der neuen DAC-Probe 32) auf 0,55 g/ml standardisiert. Weitere Substanzen (z. B. Lactose oder mikrokristalline Cellulose) können ebenfalls zum Einsatz kommen, hierzu finden sich weitere Hinweise ebenfalls im DAC/NRF.
Methode A
Methode Akommt bei einem hohen Wirkstoffanteil von über 50% des Kapselfüllvolumens zur Anwendung. Da zu befürchten ist, dass durch den Wirkstoff das Fließverhalten der gesamten Füllung zu schlecht wird, wird hierbei auch dem Wirkstoff ein Anteil von 0,5% hochdispersem Siliciumdioxid als Fließregulierungsmittel zugesetzt. Im Messzylinder wird der Wirkstoff mit Füllstoff auf das benötigte Kalibriervolumen aufgefüllt. Es erfolgen ein abschließender Mischschritt und die Abfüllung in die Kapseln.
Methode B
Bei Methode B ist aufgrund des geringeren Wirkstoffanteils kein negativer Effekt auf das Fließverhalten der Pulvermischung zu erwarten. Daher wird kein zusätzliches hochdisperses Siliciumdioxid zugesetzt. Im Messzylinder wird zur Vermeidung von Wirkstoffverlusten zunächst etwas Füllmittel vorgelegt (etwa 25% des Kalibriervolumens). Anschließend erfolgt das Auffüllen auf das Gesamtvolumen in zwei Schritten, die von einem Mischprozess unterbrochen werden: Zunächst wird auf nur etwa 80% des Kalibriervolumens aufgefüllt, gemischt, und erst dann erfolgt das endgültige Auffüllen, Mischen und Abfüllen.
Allgemeine Überlegungen
Um am Ende der Herstellung der Pulvermischung das im Messzylinder abgemessene Volumen auch tatsächlich entsprechend des Fassungsvermögens der Kapseln vorliegen zu haben, müssen die folgenden Überlegungen berücksichtigt werden:
- Nur wenn die Kapselunterteile exakt plan mit dem Kapselbrett abschließen (weder zu hoch noch zu tief), kann das ermittelte Kalibriervolumen überhaupt dem Füllvolumen entsprechen.
- Durch Erschütterungen sackt (vor allem schlecht fließendes) Pulver vergleichsweise stark in sich zusammen, sodass je nach dem Zeitpunkt der Erschütterungen zu viel oder zu wenig Pulver bereitgestellt wird.
- Die Zerkleinerung von Pulverpartikeln (z. B. durch unvorsichtiges Mischen in einer rauen Reibschale) führt meist zu einer Verschlechterung des Fließverhaltens und zu einer Vergrößerung des Schüttvolumens.
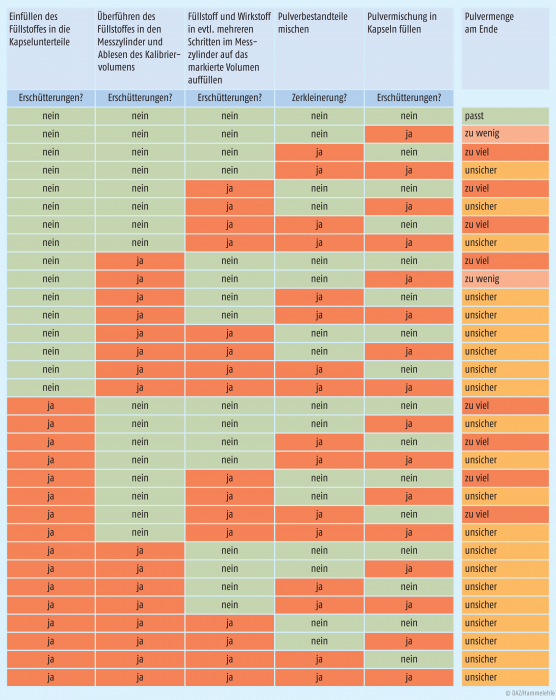
Grundvoraussetzung ist also, dass die Kapselunterteile im Kapselbrett mithilfe der Bodenplatte so justiert werden, dass eine exakte Füllung möglich ist. Geht man nun methodisch die nach Methode A und Methode B benötigten Einzelschritte der Kapselfüllung durch und überlegt, ob diese in Bezug auf Erschütterungen und Zerkleinerung fehlerfrei durchgeführt werden oder nicht, so ergibt sich das in Abbildung 3 dargestellte Bild. Es ist zu sehen, dass nur bei konsequent erschütterungsfreiem Arbeiten rein aus der theoretischen Überlegung heraus mit einem passenden Pulvervolumen zu rechnen ist. Bei den als „unsicher“ bezeichneten Ergebnissen kann nicht ausgeschlossen werden, dass ein passendes Pulvervolumen hergestellt wird – dies gilt jedoch nur, wenn sich die Fehler gegenseitig aufheben.
Befindet sich nun am Ende der Herstellung zu viel Pulver auf dem Kapselbrett, muss zunächst beachtet werden, um welche Menge es sich handelt. Ist es nur sehr wenig Pulver (und ist die benötigte Dosis relativ hoch), so kann es vertretbar sein, durch leichte Erschütterungen ein wenig Platz in den Kapseln zu schaffen und das überschüssige Pulver zu verteilen. Hierbei muss jedoch erneut aus der theoretischen Überlegung heraus klar sein: Sobald nicht exakt das Volumen des überschüssigen Pulvers freigegeben wird, kann der Überschuss nicht sicher gleichmäßig auf alle Kapseln verteilt werden. Der resultierende Fehler muss daher im Einzelfall abgeschätzt werden. Befindet sich am Ende noch vergleichsweise viel Platz in den Kapseln oder sind gar ganze Kapseln ungefüllt, so kann (wenn keine gravierenden Gegenargumente aufgrund der individuellen Herstellung vorliegen) dieser Platz mit reinem Füllstoff aufgefüllt werden. Im Anschluss müssen die Kapseln allerdings ausgeleert werden, die Gesamtfüllung ist zu durchmischen und dann erneut abzufüllen (vergleiche auch [5]).
Es darf natürlich nicht verschwiegen werden, dass Pulververluste auch durch Adsorption der Pulverbestandteile (anscheinend auch häufig selektiv des Wirkstoffes) an Messzylinder, Fanta- oder Reibschale, Pistill und Kartenblatt und auch das mit Messschritten verbundene Umfüllen auftreten. Das macht diese Methoden besonders für sehr niedrig dosierte Kapseln fehleranfällig.
Die Adsorption von Pulver an den Messzylinder kann erfahrungsgemäß durch einen Spülschritt mit wenig Aceton vor der Verwendung des Messzylinders minimiert werden. Auch ein gut fließender Kapselfüllstoff hat sich als hilfreich herausgestellt (vergleiche [1]). Mit der Ergänzungslieferung 2016/1 wurde die Qualität des zu verwendenden Mannitols definiert: Zum Einsatz kommen soll Mannitol 35, mit dem nach Mischung mit hochdispersem Siliciumdioxid direkt die vorgegebene Schüttdichte von 0,55 g/ml erreicht werden kann.
Die große Herausforderung der Herstellung einer homogenen Pulvermischung mit apothekenüblichen Methoden bleibt unabhängig von Zerkleinerungseffekten und Erschütterungen bestehen. Auch die Autorin dieser Zeilen kann noch keine abschließenden Empfehlungen aussprechen – Untersuchungen dazu laufen jedoch.
Interessant bei diesem Thema ist, dass grundsätzlich zur Herstellung niedrig dosierter Pulvermischungen die Verwendung von geometrischen Verdünnungen empfohlen wird – also das aufstockende Mischen immer gleicher Anteile aus Wirkstoff und Hilfsstoff bzw. Vormischung und weiterem Hilfsstoff [1]. Dies ist bei Arbeiten nach Methode B so jedoch nicht möglich. Es könnte daher sinnvoll sein, zunächst anteilig eine Vormischung aus Wirkstoff und Hilfsstoff herzustellen, bevor die Bestimmung des Volumens im Messzylinder erfolgt. Auch findet sich häufig die Empfehlung, den Wirkstoff in einem Bett aus Füllstoff zu umhüllen, bevor die Durchmischung erfolgt, um die Adsorption an Oberflächen zu vermindern.
Lösemethode
Für die Herstellung von Hydrochlorothiazid-Kapseln in einer Einzeldosis von 0,5 mg bis 5 mg laut NRF 26.3. ist diese Methode ein validiertes Verfahren. Die Herstellung umfasst die im Folgenden aufgeführten Schritte (wobei die entsprechenden Details dem NRF zu entnehmen sind):
- Mit Standard-Kapselfüllstoff wird das benötigte Kalibriervolumen experimentell ermittelt.
- Das abgewogene Hydrochlorothiazid wird in einer Metallfantaschale in etwas Aceton komplett gelöst.
- Etwa 40% der benötigten Füllmittelmenge wird mit der Lösung vermengt, bis kein Acetongeruch mehr wahrnehmbar ist (Verwendung von Pistill und Kartenblatt).
- Diese Mischung wird aus der Schale entnommen.
- Schale und Pistill werden mit etwas Aceton gespült, das in der Schale verbleibt.
- Mit dieser Lösung werden erneut etwa 40% des benötigten Kapselfüllstoffes vermengt, bis kein Acetongeruch mehr wahrnehmbar ist.
- Zu diesem Ansatz wird das mit dem Hauptteil des Wirkstoffes imprägnierte Füllmittel gegeben und vorsichtig vermischt.
- Im Messzylinder wird nun mit reinem Füllstoff auf das benötigte Kalibriervolumen aufgefüllt, und es erfolgt ein letzter Mischschritt vor der Abfüllung in die Kapseln.
- Nach dem Verschließen der Kapseln erfolgt eine gegenüber dem Europäischen Arzneibuch vereinfachte Prüfung auf Masseneinheitlichkeit.
Der wesentliche Vorteil dieses Verfahrens ist, dass Arzneistoffrückstände an Fantaschale und Pistill durch das Spülen mit Aceton weitgehend vermieden werden können und so die Gefahr eines Mindergehaltes erheblich geringer ist als bei herkömmlichen Methoden. Aufgrund der besonders niedrigen Dosierung an Arzneistoff (bei der absolute Gehaltsabweichungen relativ stärker ins Gewicht fallen als bei höheren Dosen) wurde diese besondere Methode gewählt.
NRF-standardisierte Methoden – Arbeiten mit Schmelzen
Hydrophile Schmelzen
Für Thalidomid-Kapseln (50 bis 200 mg) wird in NRF 32.2. das hier vorgestellte Verfahren beschrieben. Der Grund für die Vermeidung von Pulvern ist einerseits in der Arbeitssicherheit zu sehen. Andererseits handelt es sich bei Thalidomid um ein besonders unangenehm zu verarbeitendes Pulver, das sich stark elektrostatisch auflädt. Das NRF empfiehlt für die Kapseln daher die Verwendung eines Rezepturkonzentrates. Eine Bezugsquelle hierfür wird jedoch nicht empfohlen. Die Kapseln werden wie folgt hergestellt:
- Die Kapselunterteile werden in einem herkömmlichen Kapselfüllbrett zur Befüllung vorbereitet.
- Das Rezepturkonzentrat und Macrogol 4000 werden im Überschuss (10% oder weniger, je nach Kapselanzahl) auf einem siedenden Wasserbad aufgeschmolzen und verrührt. Diese Schmelze wird während der Abfüllung warmgehalten.
- Mithilfe einer weitlumigen Kanüle und einer Einmalspritze werden die Kapselunterteile vollständig (plane oder leicht konkave Oberfläche) mit der Schmelze befüllt.
- Die Füllung wird erstarren gelassen.
- Schließlich werden die Kapseln verschlossen und als In-Prozess-Kontrolle einzeln gewogen.
Lipophile Schmelzen
Diese Methode ist in der NRF-Vorschrift 22.7. zur Herstellung von Dronabinol-Kapseln (2,5 mg bis 10 mg) beschrieben. Die Vorgehensweise folgt grundlegend den folgenden Schritten:
- Die Kapselhüllen werden mithilfe eines herkömmlichen Kapselbretts geöffnet.
- Der Wirkstoff Dronabinol wird bei etwa 70 °C im Trockenschrank verflüssigt.
- Als lipophile Grundlage dient Palmitoylascorbinsäurehaltiges Hartfett – eine Stammzubereitung, die in NRF S.44. beschrieben ist. Dieses wird im Überschuss geschmolzen.
- Die angegebenen Massen des Wirkstoffes und der Grundlage werden in einem zweiten Becherglas abgewogen.
- Der Wirkstoff wird durch Rühren gelöst, und der Ansatz wird bis zum Ende der Herstellung warmgehalten.
- Mithilfe einer weitlumigen Kanüle und einer Einmalspritze werden die Kapselunterteile vollständig (plane oder leicht konkave Oberfläche) mit der Schmelze befüllt.
- Die Füllung wird erstarren gelassen.
- Schließlich werden die Kapseln verschlossen und als In-Prozess-Kontrolle einzeln gewogen.
Die Palmitoylascorbinsäure dient in dieser Zubereitung als Antioxidans für das oxidationsempfindliche Dronabinol. Das verwendete Hartfett ist kein Hartfett, wie es aus der Suppositorienherstellung bekannt ist, sondern eine streichfähige Masse. Bezugsquellen sind im NRF angegeben.
Die zähflüssige Konsistenz von Dronabinol macht diese Art der Kapselherstellung nötig, da die Verarbeitung mit einem pulverförmigen Füllstoff praktisch nicht machbar ist. Nach dieser Methode können nicht nur die beschriebenen Dosierungen hergestellt werden: Die Umrechnung auf eine andere Stärke ist aufgrund des geringen Wirkstoffanteils und damit eines angenommenen Verdrängungsfaktors von 1 einfach und mit ausreichender Genauigkeit möglich. Es ist ebenfalls möglich, wenn nötig das Kalibriervolumen der verwendeten Kapseln mit der Hartfettgrundlage selbst zu bestimmen.
Wichtige Hinweise zur Befüllung mit Schmelzen
Von der praktischen Handhabung her ist es einfacher, wenn die zu befüllenden Kapselunterteile nicht plan mit dem Kapselbrett abschließen, sondern leicht überstehen. Dies lässt sich durch die Bodenplatte bzw. die entsprechenden Muttern justieren.
Durch Volumenkontraktion entsteht beim Abkühlen ein mehr oder weniger großer Freiraum. Dieser darf allerdings keinesfalls mit Schmelze aufgefüllt werden, da dies eine Überdosierung zur Folge hätte.
Nicht durch DAC/NRF standardisierte Methoden
Ergänzungsmethode
Eine Ergänzungsmethode in Anlehnung an die Münzel-Methode zur Herstellung in Suppositorien wird verschiedentlich (beispielsweise in [2], [3], [4]) beschrieben und auch an PTA-Schulen gelehrt. Sie ist aber in der aktuellen Fassung des DAC/NRF (Stand 2016/1) nicht zu finden. Sie klingt durchaus plausibel, jedoch muss den verantwortlichen Apothekern bewusst sein, dass man sich bei ihrer Anwendung auf ungesicherterem Gebiet bewegt als bei Anwendung der herkömmlichen Methoden.
In Analogie zur Herstellung von Suppositorien nach Münzel werden die Kapseln mit einer Vormischung aus Wirk- und Füllstoff befüllt. Das Volumen dieser Vormischung darf zur vollständigen Füllung jedoch nicht ausreichen. Der leere Platz in den Kapseln wird mit reinem Füllstoff aufgefüllt. Anschließend werden die Kapseln entleert, das Pulver wird durchmischt und erneut in die Kapseln überführt.
Es kann erwartet werden, dass die Pulververluste bei dieser Methode etwas geringer sind, da kein Messzylinder benötigt wird. Doch auch hier ist äußerste Sorgfalt bei der Herstellung der Pulvermischung geboten.
Füllung mit Zuckerkügelchen
Die in DAC I.9.3.2. erwähnte Möglichkeit, Kapseln mit Zuckerkügelchen zu füllen, die in Analogie zu homöopathischen Globuli mit Wirkstoff(en) imprägniert sind, unterliegt der praktischen Limitation, dass eine Abfüllung auf Kapselbrettern nicht möglich ist: Die Kügelchen rollen unkontrolliert vom Brett (vergleiche DAC 10.3.3). Lässt sich dieses Problem lösen, sind zudem noch Validierungsstudien zu geeigneten Lösemitteln und ihrer Konzentration nötig.
Fazit
Es ist nötig, weitere Untersuchungen zur Kapselherstellung durchzuführen, um die Qualität der in Apotheken hergestellten Kapseln zu erhöhen. Ein besonderer Fokus sollte hierbei auf die Homogenität der Pulvermischung gesetzt werden, da diese unabdingbar ist, egal auf welche Weise die Abmessung der benötigten Bestandteile erfolgt. Nur für den Fall, dass kein Füllstoff benötigt wird, könnte auf den wesentlichen Mischschritt verzichtet werden – und das ist in der Praxis eben nicht der Fall.
Ansatzpunkte für diese Untersuchungen sind vor allem die Mischtechnik, die Mischdauer und das Mischgerät. In der Apotheke stehen hierbei vor allem zur Verfügung:
- raue Reibschale und Pistill (cave Zerkleinerung!)
- glatte Fantaschale und Pistill aus jeweils unterschiedlichen Materialien
- glatte Fantaschale und Kartenblatt aus jeweils unterschiedlichen Materialien
- Pulvermischdose (cave Zerkleinerung!)
- automatische Mischsysteme (z. B. Topitec®)
Bis zur Verfügbarkeit weiterer Ergebnisse muss umso mehr an den Sachverstand und das Verantwortungsbewusstsein von PTA und Apothekern appelliert werden, diese sensible Arzneiform bestmöglich herzustellen. |
Literatur
[1] Bouwman-Boer Y, Fenton-May V, Le Brun P. Practical Pharmaceutics - An International Guideline for the Preparation, Care and Use of Medicinal Products, Springer Verlag 2015
[2] Breitkreutz J, Kiefer A, Melhorn S. Fit für die Rezeptur, 2. Auflage, Govi-Verlag 2014
[3] Peuke C, Dreeke-Ehrlich M. Rezeptur für die Kitteltasche - Leitlinien für die Herstellung, 4. Auflage, Deutscher Apotheker Verlag 2013
[4] Seestädt P, Prus J, Candels T. Galenisches Praktikum für PTA, 2. Auflage, Deutscher Apotheker Verlag 2014
[5] Seidel K, Wittmann R. Rezepturaussstattung gem. ApBetrO - Geräte, Zubehör und Praxistipps, 1. Auflage, Deutscher Apotheker Verlag 2016
Autorin
Dr. Kirsten Seidel, Studium der Pharmazie in Kiel, Diplom und Promotion (2013) in der Pharmazeutischen Technologie, Schwerpunkt der Arbeiten: emulgierende Systeme. Dozentin in der Pharmazeutischen Technologie der Christian-Albrechts-Universität zu Kiel. Schwerpunkte: Herstellung steriler Arzneimittel, Arzneimittellösungen, rezepturtypische Herstellung, Biopharmazie. Mitarbeit in der Weiter- und Fortbildung von Apothekern und PTA für Rezepturthemen.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.