













- DAZ.online
- DAZ / AZ
- DAZ 52/2015
- Plasmazellen außer ...
Foto: CNRI/SPL/Agentur Focus
Medizin
Plasmazellen außer Kontrolle
Das Multiple Myelom und seine Folgen
Aufgrund ihrer unkontrollierten Proliferation bilden die Plasmazellen im Knochenmark typischerweise mehrere Tumorherde. Daraus folgt eine übermäßige Synthese monoklonaler Antikörper (Immunglobuline) durch die Plasmazellen (s. Kasten). Die Immunglobuline sind vollständig oder unvollständig und lassen sich als meist funktionslose, sogenannte Paraproteine nachweisen.
Verschiedene Formen und Vorstufen
Entsteht nur ein einzelner (solitärer) Herd, sollte man korrekterweise von einem Plasmozytom sprechen – auch wenn im klinischen Alltag die Begriffe Multiples Myelom (MM) und Plasmozytom weitgehend synonym gebraucht werden. In etwa fünf Prozent der Fälle kann sich die Erkrankung auch außerhalb eines Knochens manifestieren („extramedulläres“ Myelom); auch eine Ausschwemmung ins Blut ist möglich („Plasmazell-Leukämie“).
Als Vorstufe des MM gilt die sogenannte Monoklonale Gammopathie unklarer Signifikanz (MGUS). Der Übergang von einer MGUS zum Multiplen Myelom verläuft schrittweise und kann mehrere Jahrzehnte dauern. Ein asymptomatisches MM mit schleichendem Verlauf, bei dem bereits eine Plasmazellvermehrung im Knochenmark nachgewiesen wurde, aber noch kein Therapiebedarf besteht, nennt man „Smouldering MM“ (SMM; engl. „schwelend“).
Plasmazellen und Immunglobuline
Plasmazellen sind B-Lymphozyten im reifsten Differenzierungsstadium. Ihre Funktion ist die Synthese spezifischer Antikörper. Hierbei handelt es sich um Immunglobuline, die „ihre“ Antigene spezifisch binden. Die dadurch entstehenden Antigen-Antikörper-Komplexe werden durch das retikuloendotheliale System beseitigt.
Immunglobuline sind Proteine, die aus zwei schweren und zwei leichten Ketten bestehen; die verschiedenen Unterformen werden mit Buchstaben bezeichnet:
- bei den schweren Ketten mit A (Alpha), D (Delta), E (Epsilon), G (Gamma) und M (My),
- bei den leichten Ketten mit K (Kappa) und L (Lambda).
Jede Gruppe oder jeder Klon von Plasmazellen stellt ein bestimmtes Immunglobulin her. Ein malignes monoklonales Zellwachstum führt dementsprechend zu einer erhöhten Produktion des jeweiligen Immunglobulins. Bei etwa 80% der Patienten mit Multiplem Myelom werden vollständige Immunglobuline gebildet, wobei die Schwerkettentypen G (= IgG) und A (= IgA) am häufigsten vorkommen. So könnte in der Tumordokumentation als Proteintyp beispielsweise „IgG Kappa“ angegeben sein. In der Regel ändert sich der Proteintyp im Laufe der Erkrankung nicht.
Die pathologisch erhöhten Proteine sind – als sogenannte Paraproteine – sowohl im Blut nachweisbar als auch im Urin (hier die nach dem Erstbeschreiber benannten Bence-Jones-Proteine).
Chromosomendefekte prognostisch ungünstig
Das Multiple Myelom ist insgesamt selten, gehört aber zu den häufigsten Tumoren von Knochen und Knochenmark. Jährlich erkranken in Deutschland etwa 3000 Männer und 2700 Frauen neu, wobei die Fallzahlen der demografischen Entwicklung entsprechend zuletzt leicht zugenommen haben.
Derzeit anerkannte Risikofaktoren sind das männliche Geschlecht und ein Lebensalter über 50 Jahren. Die Pathogenese als solche ist noch weitgehend ungeklärt, verschiedene weitere kausale und/oder Einflussfaktoren werden diskutiert, vor allem
- eine chronische Infektion (z. B. Hepatitis C, HIV),
- starkes Übergewicht,
- Umweltgifte (z. B. Petrochemie), Strahlenbelastung,
- schwarzafrikanische Abstammung,
- familiäre Häufung (erhöhtes Risiko für Verwandte 1. Grades).
Besonders im Fokus stehen molekulargenetische Veränderungen. Als prognostisch ungünstige Marker gelten Chromosomenaberrationen wie
- die Translokationen t(4;14), t(14;16) und t(14;20),
- die Deletionen 17p, 13p und 1p sowie
- die Amplifikation von 1q21.
Knochen und Nieren besonders betroffen
Die pathologische Wucherung von Plasmazellen einerseits sowie die Überproduktion von Paraproteinen andererseits zeigen verschiedene deletäre Folgen. So werden im Knochenmark andere (blutbildende) Zellen verdrängt, oder es entwickeln sich allmählich Läsionen der Knochenstruktur. Weiterhin nimmt die Viskosität des Blutes zu, und mit der Zeit bilden sich Organschäden aus, vor allem der Nieren. Dennoch verläuft das Frühstadium des Multiplen Myeloms in nahezu 20 Prozent der Fälle asymptomatisch oder zumindest unspezifisch. Insgesamt kann sich eine durchaus vielgestaltige Symptomatik zeigen:
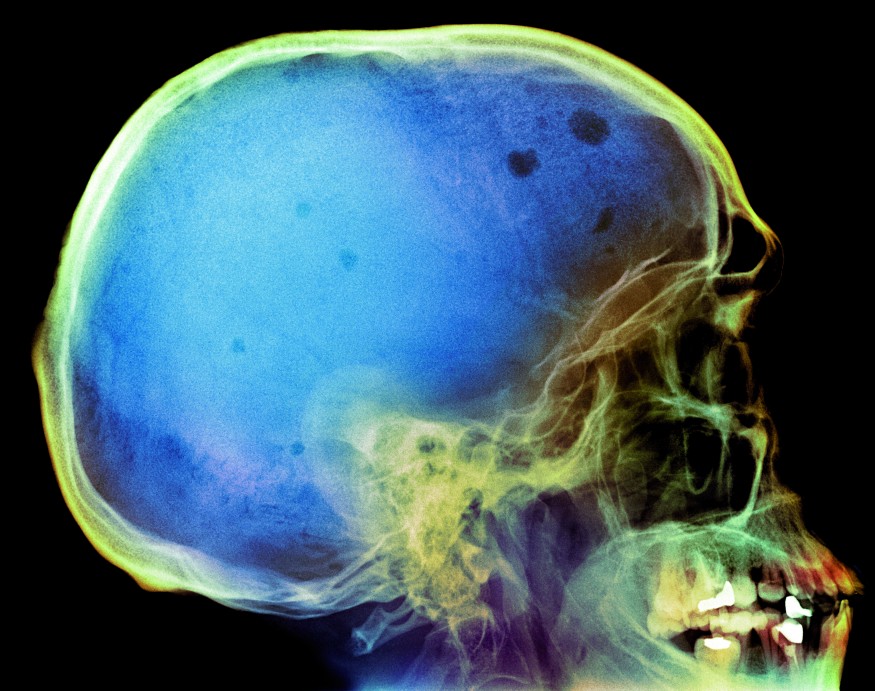
- häufig Knochenschmerzen, im weiteren Verlauf Osteoporose-Symptome wie pathologische Frakturen, Zusammensacken (Sinterung) von Wirbelkörpern, generalisierte Knochendestruktion (Osteolysen), am häufigsten an Schädel (s. Abb. 1), Rippen, Wirbelsäule, Femur, Humerus und Beckenknochen,
- oft eine anämisch bedingte Fatigue (Müdigkeit, Konzentrationsstörungen),
- oft eine Verschlechterung der Nierenfunktion mit Proteinurie (schäumender Urin),
- Gewichtsverlust bei etwa einem Viertel der Betroffenen,
- vermehrtes Auftreten von Infekten durch Leukopenie und Antikörpermangel,
- Thrombozytopenie mit Blutungen,
- Sehstörungen, Schwindel, Angina pectoris im Rahmen eines Hyperviskositätssyndroms.
Foto: Du Cane Medical Imaging Ltd/SPL/Agentur Focus
Abb. 1: Knochendestruktionen am Schädel (sog. Schrotschussschädel) eines Patienten mit Multiplem Myelom.
Folgeerkrankung Amyloidose
In etwa 15 Prozent der Fälle geht das Multiple Myelom mit einer sogenannten AL-Amyloidose einher (AL = Amyloid aus Leichtketten). Hierbei reichern sich fehlgefaltete und daher unlösliche Leichtketten-Proteine in Form von kleinen Fasern (β-Fibrillen) im Zwischenzellraum an und lagern sich dort ab. Dieser Prozess kann nahezu alle Organe betreffen, weswegen es neben den renalen auch noch zu anderen Funktionsstörungen kommen kann, beispielsweise
- kardial (Herzinsuffizienz, Arrhythmie),
- gastrointestinal (Krämpfe, Diarrhö),
- neurologisch (orthostatische Dysregulation, periphere Neuropathie).
Die AL-Amyloidose ist die häufigste der systemischen Amyloidosen und kann auch bei Patienten mit Morbus Waldenström auftreten, einem B‑Zell-Non-Hodgkin-Lymphom mit Überproduktion und Fehlfaltung von IgM.
Individuelles Therapiekonzept
Das Multiple Myelom ist nicht heilbar. Dennoch muss nicht jeder Patient mit einem positiven (Zufalls-)Befund unverzüglich behandelt werden. Sofern noch keine klinischen Symptome aufgetreten sind, wird der Erkrankungsverlauf in regelmäßigen Abständen mit Labor-, Röntgen- und Knochenmarksuntersuchungen überwacht (s. Tab. 1). Zeigen sich jedoch erste Symptome, sollten die sogenannten CRAB-Kriterien der International Myeloma Working Group (IMWG) als Entscheidungshilfe für eine Primärtherapie herangezogen werden (s. Tab. 2). Hierbei reicht bereits die Erfüllung eines einzigen Kriteriums als Behandlungsindikation aus. Als weitere Indikationen gelten Anzeichen eines Hyperviskositätssyndroms sowie einer Nierenfunktionsstörung auch bei normalen Creatininwerten.
Laboruntersuchungen |
|
Bildgebende Verfahren |
|
Zytologie / Histologie |
|
C |
„calcium“
Hyperkalzämie
|
Serum-Calcium > 2,75 mmol / l (> 10,5 mg/dl) oder > 0,25 mmol / l oberhalb des oberen Normwertes |
|---|---|---|
R |
„renal function“
Niereninsuffizienz
|
Serum-Creatinin ≥ 2,0 mg/dl (> 173 mmol / l) |
A |
„anemia“
Anämie
|
Hämoglobin < 10,0 g/dl oder ≥ 2,0 g/dl unterhalb des unteren Normwertes |
B |
„bone“
Knochen
|
Osteolyse(n), Osteoporose, Osteopenie, pathologische Fraktur |
Behandlungsziel ist, die Erkrankung über einen möglichst langen Zeitraum stabil und die Beschwerden so gering wie möglich zu halten. Daher wird für jeden Patienten das Therapieregime individuell festgelegt. Berücksichtigt werden nicht nur sein Alter und seine persönliche Lebenssituation, seine körperliche Verfassung und eventuelle Komorbiditäten, sondern gegebenenfalls auch zytogenetische Risikofaktoren.
Radio- und Chemotherapie
Bei einem solitären Herd, also einem Plasmozytom, gilt als Therapie der Wahl die kurative lokale Bestrahlung, bei Frakturen flankiert von einem chirurgischen Eingriff. Hauptziel ist, schwerwiegende Knochenzerstörungen unter Kontrolle zu bringen und die meist starken Knochenschmerzen zu lindern. Allerdings entwickeln bis zu 50 Prozent der bestrahlten Patienten im weiteren Verlauf ein Multiples Myelom.
Demgegenüber ist die Standardtherapie für ein symptomatisches Multiples Myelom eine systemische Chemotherapie. Bei Patienten unter 65 bis 70 Jahren mit noch guten Organfunktionen erfolgt zunächst eine Induktionstherapie, dann konsolidierend eine Hochdosistherapie in Verbindung mit einer Stammzelltransplantation (s. u.).
Für Patienten, die älter als 70 Jahre oder multimorbide sind, kommt in der Regel die kombinierte Hochdosis-Stammzell-Therapie nicht infrage. Hier wird eine Kombinationstherapie aus niedrigdosiertem Melphalan mit Prednisolon und Bortezomib oder Thalidomid empfohlen.
Induktionstherapie
Auch wenn verschiedene Fragen zur optimalen Dosierung, Anwendungsdauer und Kombination der Arzneimittel noch strittig sind, so ist doch klar: Die Induktionstherapie mit „neuen Substanzen“ (s. Kasten), nämlich
- dem Proteasom-Inhibitor Bortezomib (Bo),
- den Immunmodulatoren Thalidomid (T), Lenalidomid (L) und Pomalidomid und
- dem Histon-Deacetylase(HDAC)-Inhibitor Panobinostat,
zeigt bessere Ansprechraten als die klassische Vincristin-haltige Induktionstherapie, die daher nicht mehr empfohlen wird.
Je nach Regime kommen zusätzlich noch
- Dexamethason (D) und
- die Zytostatika Doxorubicin (A) oder Cyclophosphamid (C)
zum Einsatz.
Die „neuen Substanzen“ der Myelomtherapie
In den letzten Jahren haben sich neue Substanzen mit unterschiedlichen Wirkmechanismen zur Therapie des Multiplen Myeloms bewährt, sowohl in Kombination mit einer Chemotherapie als auch vor und nach einer Hochdosistherapie mit Stammzelltransplantation:
- Bortezomib (Velcade®) gehört zu den Proteasom-Inhibitoren. Proteasome wirken in der Zelle als multikatalytische Proteasen und haben eine große Bedeutung für die Regeneration von Tumorzellen. Durch ihre Hemmung werden DNA-Reparaturmechanismen in Myelomzellen blockiert, sodass deren Resistenz gegenüber Zytostatika überwunden werden kann. Als Nebenwirkung im Therapieverlauf sind schwere Polyneuropathien bei zehn bis 20% aller Patienten beschrieben. Allerdings lässt sich bei subkutaner Applikation die Rate an Polyneuropathien im Vergleich zur intravenösen Gabe signifikant senken.
- Ein neuerer Vertreter der Proteasom-Inhibitoren ist Carfilzomib (Kyprolis™, in den USA zugelassen). Bislang zeichnet sich die Substanz durch eine geringe Neurotoxizität aus und scheint Bortezomib bezüglich Wirksamkeit und Nebenwirkungen überlegen zu sein.
- Lenalidomid (Revlimid®) gehört zur zweiten Generation der Immunmodulatoren, die vom Thalidomid (früher: Contergan®) abgeleitet sind. Wichtige Wirkmechanismen sind die Antiangiogenese (Hemmung der Gefäßneubildung in Tumoren), die Hemmung der Produktion proinflammatorischer Zytokine und die Stimulation von T-Lymphozyten und natürlichen Killerzellen. Bei gleicher und besserer Wirksamkeit als Thalidomid zeigt es deutlich geringere Nebenwirkungen (Thalidomid kann Neuropathien, Fatigue und thromboembolische Komplikationen verursachen; für Frauen im gebärfähigen Alter ist es wegen seines hohen teratogenen Risikos kontraindiziert). Lenalidomid eignet sich daher auch für Myelom-Patienten, die mit Thalidomid oder Bortezomib nicht (mehr) behandelt werden können.
- Zur dritten Generation dieser Immunmodulatoren gehört Pomalidomid (Imnovid®). Es erwies sich bislang auch bei Patienten als wirksam, die trotz Vortherapien (einschließlich Lenalidomid oder Bortezomib) eine Krankheitsprogression aufwiesen.
- Mit Panobinostat (Farydak®) steht der Medizin erstmals ein Histon-Deacetylase(HDAC)-Inhibitor, also ein Wirkstoff mit epigenetischer Aktivität, zur Verfügung. Panobinostat ist zur Behandlung des Multiplen Myeloms in Kombination mit Bortezomib und Dexamethason zugelassen und in Deutschland seit Oktober 2015 auf dem Markt.
Laut DGHO-Leitlinie (Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie) sind beispielsweise BoCD, BoLD, BoAD, BoD oder BoLCD geeignete Kombinationen. Bezüglich Bortezomib konnte auch nachgewiesen werden, dass es die Prognose von Patienten mit der Translokation t(4;14) verbessert.
Bis zur Stammzellgewinnung erhält der Patient meistens drei, manchmal bis zu sechs Therapiezyklen.
Hochdosistherapie mit Stammzelltransplantation
Im Gegensatz zu den Leukämien kommt beim Multiplen Myelom in der Regel eine autologe Stammzelltransplantation zur Anwendung; dem Patienten werden also seine eigenen Stammzellen – als „Mutterzellen“ aller Blutzellen – übertragen. Diese werden zuvor in der Remissionsphase aus Blut oder Knochenmark gewonnen, nachdem sie zytotoxisch „mobilisiert“ worden sind, z. B. mit Cyclophosphamid und G-CSF (Granulozyten-Kolonie-stimulierender Faktor).
Zwischen der Stammzellgewinnung und -transplantation erhält der Patient eine systemische Hochdosis-Chemotherapie, in der Regel mit dem Zytostatikum Melphalan. Wird durch dieses Procedere keine gute Remission erreicht, empfiehlt sich eine zweite Transplantation („Tandem-Transplantation“). Auch für die Erhaltungstherapie nach Transplantation zeigten die „neuen Substanzen“ Bortezomib, Lenalidomid und Thalidomid positive Resultate in Bezug auf ein verlängertes progressionsfreies und Gesamtüberleben. Allerdings wurde bei längerfristiger Gabe von Lenalidomid und Thalidomid ein geringer Anstieg von Zweitneoplasien beobachtet.
Die einzige kurative Therapieoption beim Multiplen Myelom wäre eine allogene, also Spenderzellen-Transplantation. In mehreren Studien zeigten Patienten mit einer 17p-Deletion tatsächlich bessere Daten im Hinblick auf das Gesamt- sowie progressionsfreie Überleben, wenn auf die autologe eine allogene Stammzelltransplantation folgte.
Supportive Maßnahmen
Begleitend sollte die Lebensqualität der Patienten durch eine gezielte Behandlung von Symptomen erhöht werden. Je nach Situation kommen Analgetika und Erythropoietin zum Einsatz. Bei einem Antikörpermangelsyndrom mit häufigen Infekten kann IgG intravenös substituiert werden, bei einem Hyperviskositätssyndrom kann eine therapeutische Plasmaseparation durchgeführt werden.
Um die Aktivität der Osteoklasten und damit den Knochenabbau zu hemmen, sollte die Indikation zur Therapie mit Bisphosphonaten beim Multiplen Myelom großzügig gestellt werden. Es konnte mehrfach nachgewiesen werden, dass Bisphosphonate die Rate sogenannter Skelettereignisse (Frakturen, Zusammensinterung von Wirbelkörpern u. a.) signifikant senken können. Eine gravierende Nebenwirkung von Bisphosphonaten sind Kieferosteonekrosen, daher sollte vor Therapiebeginn ein gründliches zahnärztliches Konsil erfolgen.
Neue Strategien: aktivierte T‑Zellen und monoklonale Antikörper
Verschiedene klinische Studien der letzten Zeit befassten sich mit gentechnologischen oder abortiven Zelltherapien. So wurden bei Patienten mit therapierefraktärem Multiplem Myelom T‑Zellen aus dem Blut gewonnen und anschließend im Labor mithilfe von Lentiviren mit dem Gen für einen chimären Antigenrezeptor ausgestattet. Dieser erkennt B‑Zellen an einem bestimmten Merkmal, weswegen die aktivierten T‑Zellen nach Rückinfusion die schädlichen B‑Zellen im Körper des Patienten angreifen. Von zehn Studienteilnehmern, die sowohl eine autologe Stammzell- als auch eine T‑Zell-Therapie erhielten, befinden sich derzeit sechs in Vollremission.
Ein anderer Ansatz zur Stimulierung der T‑Zell-Antwort beruht auf der Entnahme von T‑Zellen aus dem Knochenmark („marrow infiltrating lymphocytes“, MIL), die anschließend im Labor isoliert, vermehrt und aktiviert werden. Letzteres geschieht mithilfe spezifischer Antikörper, die zwar nicht direkt gegen B‑Zellen gerichtet sind, dafür aber die Proliferation körpereigener T‑Zellen stimulieren. Bei 13 von 22 Studienteilnehmern mit neu diagnostiziertem oder rezidiviertem Plasmozytom kam es nach der MIL-Therapie zu einer partiellen Remission – bei sieben sogar zu einer Tumorreduktion um 90 Prozent; allerdings hatten die Patienten vor der MIL-Therapie auch eine Chemo- oder Stammzelltherapie erhalten.
Eine vielversprechende Strategie zur Behandlung des Multiplen Myeloms stellen die monoklonalen Antikörper dar, die bereits bei anderen malignen Erkrankungen therapeutisch fest verankert sind. So wird derzeit der Antikörper Elotuzumab – kombiniert mit Lenalidomid und Dexamethason – in Phase-III-Studien vor und nach einer Hochdosistherapie mit autologer Stammzelltransplantation für Patienten bis 70 Jahren geprüft. |
Literatur
[1] Multiples Myelom. In: Herold G, et al. Innere Medizin, 2014: 80-84
[2] Onkopedia Leitlinie Multiples Myelom. Empfehlungen der Fachgesellschaft zur Diagnostik und Therapie hämatologischer und onkologischer Erkrankungen (DGHO). September 2013
[3] Gumpp V, Engelhardt M. Multiples Myelom (Plasmozytom). Klinisches Krebsregister Universitätsklinikum Freiburg. März 2015
[4] National Comprehensive Cancer Network. Multiple Myeloma. NCCN Guidelines for Patients. Version 1.2015
[5] www.krebsgesellschaft.de > Basis-Informationen Krebs > Krebsarten > Multiples Myelom
Autor
Clemens Bilharz ist Facharzt für Anästhesie und Intensivmedizin und zusätzlich als wissenschaftlicher Fachzeitschriftenredakteur ausgebildet. Er ist als Autor und Berater für Fachverlage und Agenturen tätig.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.