













- DAZ.online
- DAZ / AZ
- DAZ 5/2015
- Gegen das Vergessen
Alzheimer-Demenz
Gegen das Vergessen
Therapeutische Strategien im Kampf gegen Morbus Alzheimer

Foto: Igor Mojzes – 123rf.com
Im Jahr 1901 beschrieb der deutsche Pathologe Alois Alzheimer erstmals die nach ihm benannte Erkrankung. Die Symptomatik einer erst 50-jährigen Patientin ging mit Orientierungslosigkeit, Gedächtnisverlust und Stimmungsschwankungen einher. Die posthume Untersuchung der Hirnrinde zeigte das Vorhandensein unphysiologischer Eiweißplaques, die als Ursache für die Neurodegeneration angesehen wurden und heute als ein typisches Kennzeichen des Morbus Alzheimer gelten.
Neben der genetischen Prädisposition zählen ein hohes Alter, vorangegangene Schädel-Hirn-Trauma, Diabetes und hohe Cholesterol-Werte sowie kardiovaskuläre Erkrankungen zu den Risikofaktoren des Morbus Alzheimer. Tatsächlich sind hiervon fast ausschließlich Personen höheren Alters betroffen, wobei sich die Prävalenz der Erkrankung zum Jahr 2050 nahezu vervierfachen wird. Diese exponenzielle Zunahme an Neuerkrankungen in der Bevölkerung stellt die heutige Medizin vor enorme Herausforderungen, da derzeit nur wenige Arzneimittel in Deutschland zugelassen sind, um die Symptomatik der Alzheimer-Krankheit zu behandeln und die Progression zu verzögern. Hierzu gehören Vertreter der Gruppe der Cholinesterase-Inhibitoren sowie der NMDA-Rezeptor-Antagonist Memantin.
Modulation der Neurotransmission
Tatsächlich spielen cholinerge- und glutamaterge Neurone eine wichtige Rolle in der Entwicklung und Erhaltung der kognitiven Leistungsfähigkeit. Der Verlust dieser Neurone gilt als anerkannter Pathomechanismus des Morbus Alzheimer, wobei die neuronalen Dysfunktionen mit einer reduzierten Aktivität des Neurotransmitters Acetylcholin und einer erhöhten extrazellulären Konzentration an Glutamat einhergehen. Die Modulation der Neurotransmission gilt daher als vielversprechender Ansatz, eine symptomatische Verbesserung zu erzielen.
Die Verwendung der Cholinesterase-Inhibitoren Donepezil (z. B. Aricept®), Galantamin (z. B. Reminyl®) und Rivastigmin (z. B. Exelon®) verhindert den Abbau von Acetylcholin im synaptischen Spalt und verstärkt somit die cholinerge Neurotransmission. Klinische Daten belegten eine Verbesserung der kognitiven Funktionen und eine verlangsamte symptomatische Progression und damit den therapeutischen Nutzen der Monotherapie gegenüber Placebo [2]. Die pharmazeutische Entwicklung fokussiert sich derzeit auf die Möglichkeit einer transdermalen Wirkstoffapplikation, um gastrointestinale Nebenwirkungen zu vermeiden. In Tierstudien wurde durch eine intranasale Administration eine höhere Bioverfügbarkeit im ZNS erreicht, als durch eine orale Applikation. Auch die direkte Modulation der muscarinergen und nicotinergen Cholinozeptoren wird derzeit evaluiert, wobei eine Vielzahl an Agonisten, Partialagonisten und allosterischen Modulatoren bereits klinisch erprobt werden.
Die Dysfunktion cholinerger Neurone ist bereits im frühen bis mittleren Stadium der Alzheimer-Erkrankung von pathologischer Bedeutung; im späteren Verlauf auch das glutamaterge System. Dabei wird eine extrazelluläre Akkumulation des Neurotransmitters beobachtet, die die dauerhafte Aktivierung postsynaptischer NMDA-Rezeptoren provoziert.
Memantin (z. B. Axura®,Ebixa®)ist derzeit das einzige zugelassene Therapeutikum zur Behandlung des Morbus Alzheimer im mittleren bis schweren Stadium. Als nicht-kompetetiver NMDA-Antagonist blockiert es den Einstrom von Calcium-Ionen durch die neuronale Membran. Wegen einer spannungsabhängigen Affinität blockiert Memantin vorzugsweise nur die pathologisch verstärkt aktiven Rezeptoren. Experimentelle Daten bestätigen zudem antagonistische Effekte auf nicotinerge Cholinozeptoren sowie serotonerge-, histaminerge und dopaminerge Neurone, sodass die Wirksamkeit von Memantin möglicherweise auch auf andere Modulationen zurückzuführen ist. Während klinische Studien und Metaanalysen den Nutzen der Monotherapie bisher ausschließlich für spätere Stadien der Krankheit bestätigten, weisen einzelne Untersuchungen auch auf synergistische Effekte in frühen Phasen der Alzheimer-Erkrankung durch Kombination mit Cholinesterase-Inhibitoren hin [3]. Eine Fixkombination aus Donepezil und Memantin befindet sich derzeit in einer klinischen Studie der Phase III. Zukünftige Untersuchungen fokussieren sich zudem auf das Potenzial einer kausalen Therapie durch Memantin, da eine mögliche Assoziation der pathologisch aktivierten NMDA-Rezeptoren mit dem vermehrten Auftreten von amyloiden Aβ-Peptiden vermutet wird. Die Bedeutung dieser toxischen Oligomere in der Pathophysiologie des Morbus Alzheimer wird in späteren Abschnitten diskutiert [4].
Nach neueren Erkenntnissen erscheint neben der Dysfunktion cholinerger und glutamaterger Neurone auch die Aktivität anderer Botenstoffe, wie γ-Aminobuttersäure (GABA), Histamin und Serotonin gestört. Neue GABA-Rezeptor-Antagonisten wie Etazolat oder SGS742 zeigten bereits neuroprotektive Wirkungen, jedoch fehlen derzeit klinische Daten zur langfristigen kognitiven Leistungssteigerung [5]. Zudem werden derzeit einige bereits zugelassene Inhibitoren der Monoaminoxidase (MAO) und selektive Serotonin-Wiederaufnahmehemmer (SSRI) aufgrund ihrer Serotonin-mimetischen Eigenschaften an Alzheimer-Patienten getestet [6]. Serotonin-Rezeptoren finden sich u. a. in Hirnregionen, die für kognitive Leistungen verantwortlich sind, wobei die Rezeptordichte bei Morbus Alzheimer pathologisch reduziert erscheint. Neuartige, direkte Serotonin-Rezeptor-Modulatoren werden derzeit in klinischen Studien der Phase I bis II als Mono- sowie Kombinationstherapeutika evaluiert [7, 8]. Auch Histamin-Rezeptoren (H3) regulieren nachweislich die kognitive Leistungsfähigkeit. Im Rahmen einer Pilot-Studie verbesserten neuartige H3-Rezeptor-Antagonisten die Gedächtnisleistung sowie die Aufmerksamkeit behandelter Alzheimer-Patienten [9].
Inwiefern die Vielzahl an symptomatischen Behandlungsstrategien tatsächlich die bisherigen Therapieoptionen ergänzen können, bleibt abzuwarten. Trotz der durchweg positiven Daten aus präklinischen Studien bzw. der Anwendung an definierten Patientenkollektiven fehlen derzeit Ergebnisse aus repräsentativen Langzeitstudien, sodass die zukünftige Bedeutung der neuartigen Wirkstoffe noch nicht abgeschätzt werden kann.
Tau-basierte Therapien
Die Alzheimer-Erkrankung gehört dem neurodegenerativen Krankheitsbild der Tauopathien an. Tau-Proteine binden unter physiologischen Bedingungen an das Zytoskelett (Mikrotubuli) der Nervenzellen und regulieren hierdurch die funktionale Organisation der Neurone, insbesondere die axonale Morphologie, Wachstum und Zellpolarität [10]. Von besonderer Bedeutung ist der Phosphorylierungsgrad des Tau-Proteins. Mit zunehmender Phosphorylierung verliert das Tau-Protein an Bindungsaffinität und verursacht so eine Instabilität der Mikrotubuli. Hirngewebe von Alzheimer-Patienten weist typische, mikroskopisch sichtbare Ablagerungen hyperphosphorylierter Tau-Proteinaggregate (neurofibrilläre Tangles) in Nerven- und Gliazellen auf. Es ist derzeit noch ungeklärt, ob diese primär die zelluläre Degeneration verursachen oder erst sekundär gebildet werden. Therapieoptionen, die gegen toxische Tau-Aggregate gerichtet sind, stellen möglicherweise erstmals rationale Ansätze dar, die Progression der Alzheimer-Erkrankung zu verhindern. Doch auch hier fehlen entsprechende Langzeitstudien, sodass deren therapeutische Relevanz derzeit noch nicht abgeschätzt werden kann.
Mit einem weiteren rationalen Therapieansatz wird versucht, die enzymatische Phosphorylierung der Proteine zu verhindern, wobei auf die Glykogen-Synthase-Kinase-3 (GSK3) als primäres Enzym der Tau-Phosphorylierung fokussiert wird. Lithium und Valproat zeigten zwar bereits inhibitorische Kapazität gegenüber GSK3 und reduzierten den Phosphorylierungsgrad der Tau-Proteine in transgenen Mäusen [11], jedoch fehlen bisher kontrollierte Studien am Menschen. Auch Coffein scheint in präklinischen Studien inhibitorische Effekte auf GSK3 zu besitzen. So weisen epidemiologische Daten auf die geringe Inzidenz der Alzheimer-Erkrankung unter starken Kaffee-Konsumenten hin. Jedoch fehlen derzeit ebenfalls kontrollierte klinische Studien [12]. Mit Tideglusib, einem irreversiblen Inhibitor der GSK3β-Isoform, konnten bereits im Rahmen einer klinischen Phase-II-Studie signifikant kognitive Funktionen verbessert werden. Eine alternative Strategie zur Erhaltung des physiologischen Phosphorylierungsgrades der Tau-Proteine liegt in deren Dephosphorylierung durch Aktivierung des Enzyms Phosphatase 2A mittels Natrium-Selenit. Dieser Ansatz wird derzeit in klinischen Studien der Phase II evaluiert.
Mit neuartigen Arzneistoffen, die sich bereits in klinischen Studien der Phase 1 befinden, wird eine direkte Stabilisierung der Mikrotubuli versucht. Methylenblau inhibiert dagegen die Tau-Tau-Interaktion und verhindert somit die pathophysiologische Ansammlung hyperphosphorylierter Tangles. Mit dem Methylenbau-Abkömmling LMTXTM (TauRx Therapeutics), der daraufhin entwickelt wurde, konnte die Progression der Alzheimer-Erkrankung über 50 Wochen stabilisiert werden. Er befindet sich nun in klinischen Studien der Phase III.
Auch natürliche Pflanzeninhaltsstoffe, wie das Oleuropein bzw. dessen Aglykon aus der Olive scheinen ähnlich inhibierende Eigenschaften zu besitzen [13]. Ebenso steht die Spaltung bereits gebildeter Tangles im Fokus der Forschung. Das Hitzeschockprotein Hsp90 vermittelt die Faltung der Aggregate und schützt diese so vor enzymatischer Spaltung. Inhibitoren der Hsp90 Chaperone, wie Curcumin und EC102 konnten in Tierversuchen bereits die Tau-Akkumulation verringern [14]. Letztlich wird auch versucht, das körpereigene Immunsystem in Tau-basierte Therapien zu involvieren. Im Rahmen passiver Vakzinierungen mittels monoklonaler Antikörper gegen Tau-Oligomere zeigten sich verbesserte motorische Leistungen in transgenen Mäusen [15].
Amyloid-basierte Therapien
Das β-Amyloid (Aβ) entsteht nach enzymatischer Spaltung des Amyloid-Precursor-Proteins (APP) durch α-, β- und γ-Sekretasen. Aβ-Peptide werden unter physiologischen Bedingungen kontinuierlich erzeugt und besitzen unter anderem antimikrobielle Eigenschaften. Während die α-/γ-Sekretasen nicht-amyloide Peptide erzeugen, führt das alternative Spalten durch die β-/γ-Sekretasen zur Bildung neurotoxischer Aβ-Peptide. Diese sind Hauptbestandteil der amyloiden Plaques und gelten neben den Tau-Aggregaten als typische Kennzeichen des Morbus Alzheimer. Rationale Therapieansätze zielen daher auf die Aktivierung der α-Sekretasen, um die Bildung toxischer Produkte zu verhindern. Die zukünftige Relevanz der Amyloid-basierten Therapie kann aber derzeit noch nicht abgeschätzt werden.
Bryostatin 1 sowie das Polyphenol Epigallocatechin-gallat, eines der Hauptinhaltsstoffe des Grünen Tees, gelten als solche Induktoren [16]. Ihre Wirksamkeit in frühen Stadien der Erkrankung wird derzeit in klinischen Phase-II-Studien evaluiert. Auch gibt es bereits einige neuartige Substanzen mit potenziell inhibitorischem Potenzial gegen β-Sekretasen, die in präklinischen sowie klinischen Phase-I- und -II-Studien untersucht werden [17].
Neben der Synthese amyloider Peptide gilt auch deren Transport durch die Blut-Hirn-Schranke als mögliches therapeutisches Target [18]. Hierbei vermittelt das Protein LRP (Low-density lipoprotein receptor-related protein) den Transport des Aβ-Peptids über die Blut-Hirn-Schranke und somit dessen Abtransport in die Peripherie. Da mit zunehmendem Alter die Expression dieses Transporters abnimmt, verlängert sich die Verweildauer des toxischen Aβ-Peptids im ZNS und das Risiko neurotoxischer Effekte steigt. Derzeit steht die Entwicklung rekombinanter LRP-Transporter im Fokus der Forschung. Demgegenüber erleichtert der RAGE (Receptor for advanced glycation end products)-Rezeptor die Aufnahme von Aβ-Peptiden in das ZNS. Alzheimer-Patienten zeigen eine erhöhte Expression dieses Proteins, sodass kompetitive Inhibitoren des Rezeptors bereits in klinischen Phase-I- und –II-Studien getestet werden [19].
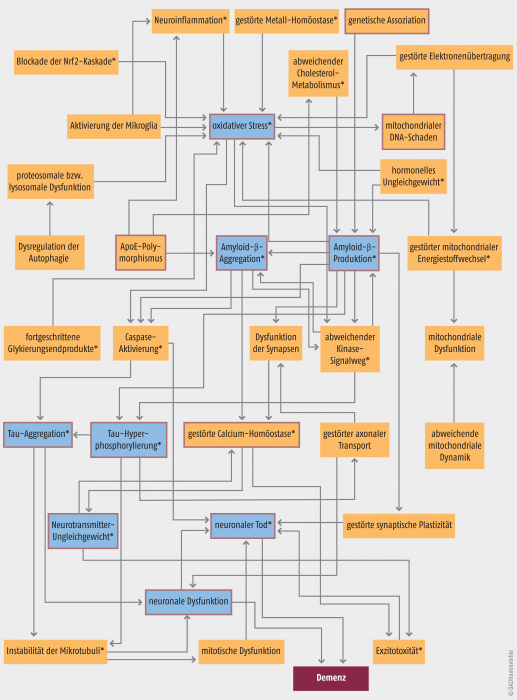
Pathogenese der Alzheimer-Demenz. Die Abbildung zeigt die vielen verschiedenen Mechanismen, die bei der Pathogenese der Demenz eine Rolle spielen. Die wichtigsten sind fett farbig umrahmt. Gegen die mit * markierten Mechanismen werden bereits Arzneistoffe entwickelt. (Quelle: [1] Anand R et al. Therapeutics of Alzheimer‘s disease: Past, present and future. Neuropharmacology 2014;76A(0):27-50)
Das Glucosaminoglykan Tramiprosat, das mit hoher Affinität Aβ-Peptid-Monomere bindet und somit deren Aggregation zu toxischen Oligomeren verhindert, stellt eine weitere Therapieoption dar. Die Wirksamkeit von Tramiprosat wurde bereits in einer großen klinischen Phase-III-Studie untersucht. Es konnte jedoch keine signifikante Verbesserung der primären Endpunkte erreicht werden. Dennoch zeigten Subanalysen leichte kognitive Verbesserungen bei den Probanden und auch die neuronale Atrophie reduzierte sich signifikant [20]. Daneben sind auch einige Enzyme wie Plasmin, Neprilypsin und Angiotensin-Konversions-Enzym (ACE) in der Lage, toxische Aβ-Aggregate zu spalten und hierdurch die Menge an amyloiden Plaques zu reduzieren. Daher gelten auch spezifische Enzymaktivatoren als rationale Therapieoption. Bisher deuten derzeit ausschließlich präklinische Daten auf eine klinische Wirksamkeit.
Auch existieren erste passive Immunisierungsversuche mit monoklonalen Antikörpern (Bapineuzumab, Solanezumab und Gantenerumab) gegen amyloide Aβ-Peptide. Trotz vielversprechender präklinischer Daten konnten in klinischen Phase-III-Studien keine signifikanten Therapie-relevanten Erfolge beobachtet werden. Derzeit stehen in klinischen Phase-II-Studien Versuche der aktiven Immunisierung mittels Anti-Amyloid-Vakzinen im Fokus der Forschung.
Neuere Studien zeigten, dass Aβ-Peptide direkt die intrazellulären Signalkaskaden der Zellkommunikation modulieren und hierdurch synaptische Defizite vermitteln. cAMP sowie Stickstoffmonoxid-(NO)-abhängige Proteinkinasen wurden hierbei als Effektormoleküle identifiziert. Die Inhibition der Phosphodiesterase mittels Rolipram, Sildenafil und Cilostazol verbesserte in ersten experimentellen Tierversuchen die kognitive Gedächtnisleistung und verringerte neuronale Schäden. Klinische Daten am Menschen fehlen derzeit aber noch.
Behandlung des oxidativen Stresses
Oxidativer Stress gilt als wichtiger pathogener Faktor in der Entstehung des Morbus Alzheimer. Diese unphysiologische Stoffwechsellage geht mit der vermehrten Bildung reaktiver Sauerstoffspezies (ROS) einher und führt zur Schädigung der zellulären Organellen. Neue Therapiestrategien zielen daher auf eine Minimierung oxidativer Einflüsse. Eine einfache Möglichkeit, radikale Sauerstoffspezies abzufangen, sind exogene Antioxidanzien wie Vitamin C, E und Carotinoide sowie phytochemische Substanzen (Polyphenole und Flavonoide). Vitamin E reduzierte zwar die Lipid-assoziierte Peroxid-Bildung in transgenen Mäusen, zeigte am Menschen jedoch keine Therapie-relevanten Vorteile, weder als Monotherapie noch in Kombination mit Donepezil [21]. Auch Resveratrol, ein Phytoalexin aus Weintrauben, verbesserte die Verhaltensweise transgener Mäuse, wobei auch epidemiologische Daten darauf verweisen, dass moderater Weingenuss das Demenzrisiko vermindert [22].
Melatonin, das als Hormon der Zirbeldrüse den Tag-Nacht-Rhythmus des menschlichen Körpers steuert, besitzt ebenso antioxidative Eigenschaften. Auch andere pleiotrope Effekte, welche die Progression von Morbus Alzheimer positiv beeinflussen können, werden dem Melatonin zugeschrieben. Hierzu gehören die Inhibition der Aβ-Produktion und -Aggregation zu amyloiden Fibrillen, die Verminderung der Tau-Hyperphosphorylierung sowie antiapoptotische Effekte [23]. Klinische Studien am Menschen bestätigten bereits verbesserte kognitive und neuropsychiatrische Leistungen [24]. Die Wirksamkeit des Melatonins in späten Phasen der Erkrankung wird derzeit in klinischen Studien der Phase II evaluiert. Auch die Wirksamkeit direkter Melatonin-Rezeptor-Agonisten wird derzeit untersucht.
Problematisch ist dabei, dass die geringe zentralnervöse Bioverfügbarkeit, die rasche Metabolisierung sowie die unterschiedliche intrazelluläre Konzentration von Melatonin eine Anreicherung in Mitochondrien – dem Hauptbildungsort reaktiver Sauerstoffspezies – und damit die therapeutische Wirksamkeit verhindert. Eine Möglichkeit, diese Einflussfaktoren zu umgehen, besteht in dem Versuch, die endogenen antioxidativen Mechanismen zu aktivieren. Die zelluläre Nrf2/ARE-Kaskade (Nuclear receptor factor2/antioxidant response element) induziert nach Aktivierung die Translokation der entsprechenden Signalmoleküle in den Zellkern. Die folgende Regulation der Gentranskription ist bei Alzheimer-Patienten nachweislich gestört und provoziert vermutlich die unphysiologische Stoffwechsellage [25]. Eine Aktivierung der Kaskade durch Tert-Butyl-Hydrochinon oder CDDO-Methylamid zeigte bereits protektive Effekte gegenüber toxische Aβ-Peptide sowie der Bildung reaktiver Sauerstoffspezies.
Während diese Maßnahmen mehrheitlich die Neutralisation der gebildeten Radikale fokussieren, stellt die Modulation ihrer Bildung in den Mitochondrien eine effektivere Therapieoption dar. Das Coenzym Q10 ist hierbei zentraler Elektronenüberträger, sodass dessen Supplementierung entsprechendes neuroprotektives Potenzial besitzt. Obwohl diese Annahme durch präklinische Daten bestätigt wurde, zeigte sich in klinischen Anwendungen am Menschen kein signifikanter Therapie-relevanter Vorteil, weder als Monotherapie noch in Kombination mit weiteren Wirkstoffen. Auch Acetyl-L-Carnithin und die R-α-Liponsäure bestätigten in tierexperimentellen Studien ihre antioxidative Wirksamkeit [26]. Derzeit werden hierzu ebenfalls klinische Studien der Phase I und II durchgeführt. Inwiefern die Modulation des mitochondrialen Stoffwechselsystems tatsächlich zu einer Verbesserung kognitiver und neuropsychologischer Leistungen führt, bleibt jedoch vorerst abzuwarten.
Regulation der zellulären Calcium-Homöostase
Calium-Ionen spielen bei kognitiven Lern- und Gedächtnisprozessen eine wichtige Rolle und regulieren zudem die Viabilität der Neurone. Alzheimer-Patienten weisen eine gestörte Ca2+-Homöostase auf, was eine neuronale Dysfunktion begünstigt. Memantin, als bereits zugelassenes Medikament gegen Morbus Alzheimer, verringert den überschießenden Calcium-Ionen-Influx durch Antagonisierung des NMDA-Rezeptors und verhindert somit die toxischen Effekte einer erhöhten Ionenkonzentration. Andere Therapieansätze fokussieren sich auf die mitochondriale Calcium-Homöostase. Minocyclin und Arzneistoffe aus der Gruppe der nicht-steroidalen Antirheumatika (NSAR) inhibierten bereits in tierexperimentellen Studien die Calcium-Ionen-Aufnahme in die Zellorganellen durch Depolarisation der mitochondrialen Membran [27, 28].
Antientzündliche Therapien
Auch neuroinflammatorische Prozesse gehören zu den bekannten Pathomechanismen des Morbus Alzheimer. Sie induzieren den Zelltod der Neurone. Eine entzündungshemmende Behandlungsstrategie gilt daher als rationaler Therapieansatz. Aufgrund der bereits erwähnten Effekte auf die mitochondriale Calcium-Homöostase stellen nicht-steroidale Antirheumatika (NSAR) die bevorzugte Substanzklasse dar. Darüber hinaus sind auch modulierende Effekte auf die Rho-GTPase und damit auf das Axon-Wachstum, die Tau-Phosphorylierung und die Astrozytenmotilität bekannt und bestätigen den Einsatz von NSAR bei Morbus Alzheimer [29 – 31]. Epidemiologische Studien verweisen bereits auf die geringe Alzheimer-Inzidenz bei NSAR-behandelten Personen, jedoch konnten klinische Studien ihren therapeutischen Nutzen bisher nicht bestätigen.
Andere Behandlungsstrategien
Einige Studien berichteten bereits über die neuroprotektive Wirkung der Sexualhormone Testosteron, Estrogen und Progesteron, deren Konzentration sich altersabhängig verändert und hierdurch die Entwicklung von Morbus Alzheimer fördern kann. Hormonersatztherapien mit Gonadotropin-releasing-Hormon(GnRH)-Agonisten (z. B. Leuporelin) konnten in tierexperimentellen Studien die Abnahme der kognitiven Leistungsfähigkeit verlangsamen und die Menge an toxischen Aβ-Aggregaten reduzieren. Auch klinische Phase-II-Studien bestätigten die Zunahme der kognitiven Leistung bei weiblichen Alzheimer-Patienten. Ergebnisse einer ersten Studie der Phase III stehen jedoch noch aus.
Auch eine pathophysiologische Assoziation von Hypercholesterolämien und der Entstehung von Morbus Alzheimer wurde bereits seit Längerem vermutet. Die effektive Reduktion erhöhter Cholesterol-Spiegel mittels Statinen zeigte in experimentellen Studien zwar neuroprotektive Wirkung und positive Effekte auf die kognitive Leistungsfähigkeit [32], jedoch enttäuschten die bisherigen Ergebnisse aus klinischen Studien am Menschen.
Neurotrope Faktoren wie der spezifische Nervenwachstumsfaktor (NGF) vermitteln die Stabilisierung und Viabilität von Nervenfasern und besitzen somit das Potenzial, deren Degeneration zu verhindern. Neuartige, hochentwickelte Gentherapien werden derzeit in klinischen Phase-I- und –II-Studien getestet. Weiterhin wirkt das neuartige Peptid Cerebrolysin® (Ever Neuro Pharma) neurotrop und zeigte synergistische Effekte in Kombination mit Cholinesterase-Inhibitoren [33]. Trotz dieser vielversprechenden Resultate ist die Applikation neurotroper Faktoren auch mit der Gefahr einer anormalen Neurogenese assoziiert und bedarf daher besonderer Vorsicht [34].
Die bereits diskutierte Dysregulation der neuronalen Calcium-Homöostase ist bei Alzheimer-Patienten auch für andere Ionen beobachtet worden. Einige sind an zellulären Redoxreaktionen beteiligt, wodurch oxidativer Stress provoziert werden kann [35]. Die Administration ZNS-gängiger Chelatbildner könnte demnach hilfreich sein, die zellschädigenden Effekte zu reduzieren. Clioquinol komplexiert Zink- und Kupfer-Ionen und konnte in präklinischen Studien die Aggregation amyloider Aβ-Peptide inhibieren, jedoch erzielte die Anwendung am Menschen zunächst keine Verbesserung der kognitiven Leistungsfähigkeit.
Auch epigenetische Modifikationen, wie DNA-Methylierungen sowie Histon-Acetylierungen und die damit verbundene Veränderung der zellulären Genregulation scheinen die Entstehung neurodegenerativer Erkrankungen zu beeinflussen. Die Regulation der epigenetischen Modifikationen erfolgt hierbei durch Enzyme, deren Funktion an die Verfügbarkeit essenzieller Kofaktoren (Folsäure, Vitamin B12 und B6) gekoppelt ist. Die Supplementierung dieser Kofaktoren sowie die Modulation der entsprechenden Enzyme Histon-Acetyltransferase und Histon-Deacetylase führte bisher jedoch zu keiner Verbesserung der kognitiven Leistungsfähigkeit.
Zusammenfassung
- Das klinische Bild des Morbus Alzheimer ist durch pathologische Veränderungen einer Vielzahl zellulärer Prozesse begründet. Obwohl bedeutende molekulare Mechanismen identifiziert wurden, die mit der Pathogenese assoziiert scheinen, sind nur wenige Arzneistoffe zur symptomatischen Behandlung zugelassen. Angesichts der Vielzahl anfänglich vielversprechender präklinischer Daten, überrascht die meist nur unzureichende Wirksamkeit an Alzheimer-Patienten. Dies gilt vor allem für kausale Therapiemaßnahmen gegen hyperphosphorylierte Tau-Proteine, amyloide Aβ-Oligomere oder die Anwendung antioxidativer Substanzen. Offensichtlich sind die pathophysiologischen Mechanismen des Morbus Alzheimer noch immer nicht vollständig verstanden.
- Die besondere Lage der therapeutischen Targets im ZNS erfordert eine ausreichende zentrale Bioverfügbarkeit der Arzneistoffe.
- Einige der neuartigen Substanzen weisen pleiotrope Wirkungen auf, wodurch zum Teil gegensätzliche Effekte vermittelt werden.
- Die therapeutische Wirksamkeit scheint auch stark abhängig vom Stadium der Erkrankung zu sein. Dadurch wird der Einsatz der potenziellen Arzneistoffe auf frühe, mittlere oder späten Phasen der Alzheimer-Krankheit begrenzt.
- Faktoren wie interindividuelle genotypische Diversitäten oder fehlende Biomarker erschweren es, die Wirksamkeit der Therapieregime zu belegen.
- Vielversprechend erscheint die Möglichkeit, mit neuartigen Gentherapien die komplexen krankheitsbedingten Veränderungen auf molekularer Ebene zu beheben und zukünftig möglicherweise bereits präventiv eingreifen zu können.
Stickstoffmonoxid ist als zellulärer Transmitter an kognitiven Funktionen im ZNS, an der Regulation der Schlaf-Wach-Rhythmik und des Appetits sowie der Anpassung der Körpertemperatur beteiligt. Neuroprotektive Effekte werden zudem durch die Inhibition von Caspasen sowie die Interaktion mit NMDA-Rezeptoren vermittelt, wobei wiederum zu hohe Konzentrationen an NO neurotoxisch wirken. Auch weisen die dazugehörenden NO-Synthasen keine einheitlichen Effekte an der neuronalen Homöostase auf und besitzen neuroprotektives aber auch pro-apoptotisches Potenzial. Ein tatsächlicher Therapie-relevanter Vorteil der Modulation des NO-Systems zur Behandlung des Morbus Alzheimer wurde noch nicht bestätigt.
Weitere Behandlungsoptionen neurodegenerativer Erkrankungen sind neuartige DNA-basierte Therapeutika. Diese umfassen die zielgerichtete Applikation neurotroper Faktoren mittels Plasmid-DNA sowie die Inhibition inflammatorischer Mediatoren durch entsprechende Antisense-Oligodesoxynukleotide. Da sich diese Therapieoptionen derzeit jedoch hauptsächlich in präklinischen und klinischen Phase-I- und -II-Studien befinden, kann ihr tatsächlicher therapeutischer Nutzen bisher noch nicht abschließend bewertet werden.
Die Komplexität des pathophysiologischen Geschehens führte letztlich zu der Annahme, dass die gleichzeitige Modulation mehrerer potenzieller Faktoren eine höhere therapeutische Wirksamkeit in der Behandlung des Morbus Alzheimer besitzt als die isolierte Betrachtung einzelner Targets [36]. Die neuartige Substanz Ladostigil besitzt antioxidative Eigenschaften sowie duale inhibitorische Effekte gegen die Cholinesterase sowie Monoaminoxidase. Hierdurch konnten in präklinischen Studien bereits extrapyramidale Symptome verbessert, depressive Symptome verringert und neuroprotektive Effekte vermittelt werden [37]. Ladostigil wird derzeit in klinischen Phase-II-Studien untersucht. |
Literatur
[1] Anand R et al. Therapeutics of Alzheimer‘s disease: Past, present and future. Neuropharmacology 2014;76A(0):27-50
[2] Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006:CD005593
[3] Atri A, Molinuevo, JL, Lemming O, Wirth Y, Pulte I, Wilkinson D. Memantine in patients with Alzheimer’s disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimers Res Ther. 2013;5:6
[4] Dinamarca MC, Rios JA, Inestrosa NC. Postsynaptic receptors for amyloidbeta oligomers as mediators of neuronal damage in Alzheimer’s disease. Front Physiol. 2012:3;464
[5] Vellas, B, Sol O, Snyder PJ, Ousset PJ, Haddad R, Maurin M, Lemarie JC, Desire L, Pando MP. EHT0202 in Alzheimer’s disease: a 3-Month, randomized, placebo-controlled, double-blind study. Curr. Alzheimer Res. 2011:8;203e212
[6] Rodriguez JJ, Noristani HN, Verkhratsky A. The serotonergic system in ageing and Alzheimer’s disease. Prog. Neurobiol. 20129:9;15e41
[7] Patat A, Parks V, Raje S, Plotka A, Chassard D, Le Coz F. Safety, tolerability, pharmacokinetics and pharmacodynamics of ascending single and multiple doses of lecozotan in healthy young and elderly subjects. Br J Clin Pharmacol. 2009:67;299e308
[8] Maher-Edwards G, Dixon R, Hunter J, Gold M, Hopton G, Jacobs G, Williams P. SB-742457 and donepezil in Alzheimer disease: a randomized, placebocontrolled study. Int J Geriatr Psychiatry 2011:26;536e544
[9] Nathan PJ, Boardley R, Scott N, Berges A, Maruff P, Sivananthan T, Upton N, Lowy MT, Nestor PJ, Lai R. The safety, tolerability, pharmacokinetics and cognitive effects of GSK239512, a selective histamine H3 receptor antagonist in patients with mild to moderate Alzheimer’s disease: a preliminary investigation. Curr Alzheimer Res. 2013:10;240e251
[10] Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev. 2000:33;95e130
[11] Engel T, Goni-Oliver P, Lucas JJ, Avila J, Hernandez F. Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem. 2006:99;1445e1455
[12] Eskelinen MH, Ngandu T, Tuomilehto J, Soininen H, Kivipelto M. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009:16;85e91
[13] Daccache A, Lion C, Sibille N, Gerard M, Slomianny C, Lippens G, Cotelle P. Oleuropein and derivatives from olives as Tau aggregation inhibitors. Neurochem Int. 2011:58;700e707
[14] Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci USA 2007:104;9511e9516
[15] Lasagna-Reeves CA, Castillo-Carranza DL, Jackson GR, Kayed R. Tau oligomers as potential targets for immunotherapy for Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2011:8;659e665
[16] Smith A, Giunta B, Bickford PC, Fountain M, Tan J, Shytle RD. Nanolipidic particles improve the bioavailability and alpha-secretase inducing ability of epigallocatechin-3-gallate (EGCG) for the treatment of Alzheimer’s disease. Int J Pharm. 2010:389;207e212
[17] Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, Tang J. Beta-secretase inhibitor GRL-8234 rescues agerelated cognitive decline in APP transgenic mice. FASEB J 2011:25;775e784
[18] Fan J, Donkin J, Wellington C. Greasing the wheels of Abeta clearance in Alzheimer’s disease: the role of lipids and apolipoprotein E. Biofactors 2009:35;239e 248
[19] Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011:25;206e212
[20] Gauthier S, Aisen PS, Ferris SH, Saumier D, Duong A, Haine D, Garceau D, Suhy J, Oh J, La W, Sampalis J. Effect of tramiprosate in patients with mild-to-moderate Alzheimer’s disease: exploratory analyses of the MRI subgroup of the Alphase study. J. Nutr. Health Aging 2009:13;550e557
[21] Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev 2012:11;CD002854
[22] Orgogozo JM, Dartigues JF, Lafont S, Letenneur L, Commenges D, Salamon R, Renaud S, Breteler MB. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris) 1997:153;185e192
[23] Wang JZ, Wang ZF. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol. Sin 2006:27;41e49
[24] Cardinali DP, Furio AM, Brusco LI. Clinical aspects of melatonin intervention in Alzheimer’s disease progression. Curr Neuropharmacol. 2010:8;218e227
[25] Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007:66;75e85
[26] Siedlak SL, Casadesus G, Webber KM, Pappolla MA, Atwood CS, Smith MA, Perry G. Chronic antioxidant therapy reduces oxidative stress in a mouse model of Alzheimer’s disease. Free Radic Res. 2009:43;156e164
[27] Garcia-Martinez EM, Sanz-Blasco S, Karachitos A, Bandez MJ, Fernandez-Gomez FJ, Perez-Alvarez S, de Mera RM, Jordan MJ, Aguirre N, Galindo MF, Villalobos C, Navarro A, Kmita H, Jordan J. Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem Pharmacol. 2010:79;239e250
[28] Sanz-Blasco S, Valero RA, Rodriguez-Crespo I, Villalobos C, Nunez L. Mitochondrial Ca2þ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 2008:3;e2718
[29] Fu Q, Hue J, Li S. Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J Neurosci. 2007:27;4154e4164
[30] Sayas CL, Moreno-Flores MT, Avila J, Wandosell F. The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like Tau phosphorylation. J Biol Chem. 1999:274;37046e37052
[31] Lichtenstein MP, Carriba P, Baltrons MA, Wojciak-Stothard B, Peterson JR, Garcia A, Galea E. Secretase-independent and RhoGTPase/PAK/ERKdependent regulation of cytoskeleton dynamics in astrocytes by NSAIDs and derivatives. J Alzheimers Dis. 2010:22;1135e1155
[32] Li L, Cao D, Kim H, Lester R, Fukuchi K. Simvastatin enhances learning and memory independent of amyloid load in mice. Ann Neurol. 2006:60;729e739
[33] Allegri RF, Guekht A. Cerebrolysin improves symptoms and delays progression in patients with Alzheimer’s disease and vascular dementia. Drugs Today (Barc) 2012:48(Suppl. A);25e41
[34] Fumagalli F, Molteni R, Calabrese F, Maj PF, Racagni G, Riva MA. Neurotrophic factors in neurodegenerative disorders: potential for therapy. CNS Drugs 2008:22;1005e1019
[35] Bondy SC, Guo-Ross SX, Truong, AT. Promotion of transition metalinduced reactive oxygen species formation by beta-amyloid. Brain Res. 1998:799,91e96
[37] Youdim MB, Buccafusco JJ. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol Sci. 2005:26;27e35
[38] Youdim MB, Weinstock M. Molecular basis of neuroprotective activities of rasagiline and the anti-Alzheimer drug TV3326 [(N-propargyl-[3R)aminoindan- 5-YL)-ethyl methyl carbamate]. Cell Mol Neurobiol. 2001:21;555e573
Autor
Dr. André Said studierte von 2004 bis 2008 Pharmazie an der FU Berlin. Die Approbation als Apotheker erhielt er 2010 und arbeitete anschließend als Doktorand im Fachbereich Pharmakologie/Toxikologie an der FU Berlin bei Prof. Dr. Günther Weindl. Seine Promotion mit dem Titel: „Funktionelle Charakterisierung von dendritischen Zellen unter entzündlichen Bedingungen in vitro und Integration in humane Vollhautäquivalente“ schloss er 2014 erfolgreich ab. Seit 2013 ist Dr. André Said für die DAZ als freier Autor tätig.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.