














- DAZ.online
- DAZ / AZ
- DAZ 16/2014
- Ein Angriffspunkt, viele ...
Psychopharmaka
Ein Angriffspunkt, viele Wirkungen
Benzodiazepine, Z-Substanzen und das zentrale Nervensystem
Forschung und Zufall – die Entdeckung
Die Geschichte der Benzodiazepine hat ihren Ursprung in den 1950er Jahren. Leo Henryk Sternbach, damals Mitarbeiter der in den USA ansässigen Forschungslaboratorien der Baseler Firma Hoffmann-La Roche, entdeckte die beruhigende Wirkung des Chlordiazepoxids. Wie so oft spielte dabei der Zufall eine entscheidende Rolle. Nach zahlreichen Versuchen, Benzhexodiazine strukturell abzuwandeln und dadurch Substanzen mit einem neuartigen pharmakologischen Wirkprofil zu erhalten, wurde nach einer längeren Unterbrechung der Forschungsarbeiten eines der Derivate, das Sternbach wegen seiner besonders schönen Kristallbildung aufgefallen war, pharmakologisch getestet. Der leitende Pharmakologe, Lowell Randall, berichtete bald darauf von im Tierexperiment an Mäusen beobachteten beruhigenden, krampflösenden und muskelrelaxierenden Eigenschaften, gepaart mit einer erstaunlich großen therapeutischen Breite. Dieses Wirkprofil ist heute noch kennzeichnend für die gesamte Stoffklasse. Chlordiazepoxid wurde unter dem Handelsnamen Librium® 1960 in den USA und in Europa eingeführt und eroberte in kurzer Zeit den Markt. Noch viel größere Verbreitung fand das zweite von Sternbach entwickelte Benzodiazepin: Diazepam (Valium®).
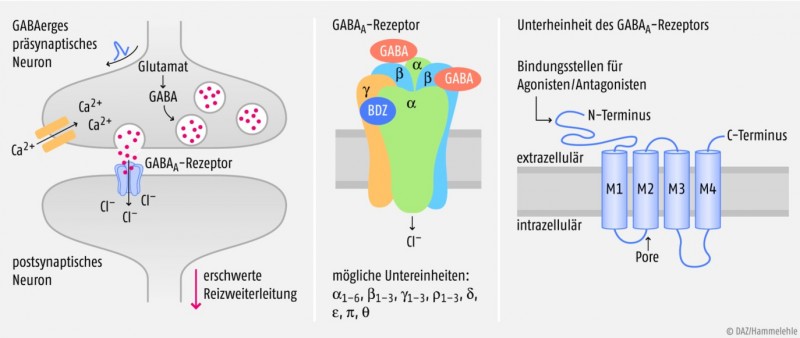
GABAA-Rezeptor und allosterische Modulation
1977 haben zwei Forschergruppen in Dänemark und in der Schweiz unabhängig voneinander „Benzodiazepin-Rezeptoren“ im Gehirn nachgewiesen. Man nahm zunächst an, dass diese „Rezeptoren“ in enger Nachbarschaft zu Ionenkanälen liegen, die durch den hemmenden Neurotransmitter GABA gesteuert werden. Heute wissen wir, dass es für Benzodiazepine keinen eigenen Rezeptor gibt, sondern dass deren Bindungsstelle zwischen zwei Untereinheiten des sogenannten GABAA-Rezeptors liegt, eines ligandengesteuerten Chloridkanals.
Am häufigsten sind im zentralen Nervensystem GABAA- Rezeptoren aus zwei α-, zwei β- und einer γ-Untereinheit zu finden. Man kennt heute beim Menschen jedoch 19 verschiedene GABAA-Untereinheiten, über welche die physiologische Aktivität des Chloridkanals bestimmt wird (Abb. 1). Diese enorme Vielfalt ist eine Erklärung dafür, dass Benzodiazepine nur in bestimmten Regionen des ZNS Wirkung zeigen.
Die Auswirkung, die ein Öffnen der GABA-regulierten Chloridkanäle auf das Membranpotenzial hat, hängt von der Ionenkonzentration in der Zelle und in ihrer Umgebung ab: In der Regel wird die Zelle beim Öffnen dieser Ionenkanäle hyperpolarisiert, d.h. Chlorid-Ionen strömen in die Zelle ein, das Membranpotenzial wird negativer und die Weiterleitung eines elektrischen Signals wird erschwert.
Der natürliche Ligand des GABAA-Rezeptors ist, wie der Name bereits sagt, γ-Aminobutyrat (GABA). Die volle Kanalaktivierung findet statt, wenn zwei GABA-Moleküle unter Beteiligung der jeweiligen α-Untereinheit an die β-Untereinheiten binden (sog. kooperative Bindung, siehe Abb. 1). Benzodiazepine können durch Bindung an die α- und γ-Untereinheit den Effekt von GABA verstärken, indem sie die Affinität von GABA zum Rezeptor erhöhen; sie sind also allosterische Modulatoren. Wichtig ist, dass die Wirkung nur auftritt, wenn gleichzeitig GABA vorhanden ist. Außerdem können die Chloridkanäle durch Benzodiazepine nie stärker aktiviert werden, als dies durch den physiologischen Liganden in hoher Konzentration möglich ist.
Der GABAA-Rezeptor besteht aus fünf Untereinheiten, die teilweise mehrere Subtypen haben; derzeit sind insgesamt 19 Isoformen bekannt. Nachdem GABA (rote Pünktchen) aus dem präsynaptischen Neuron freigesetzt worden ist, bindet es an die α- und β-Untereinheit des GABAA-Rezeptors. Benzodiazepine (BDZ) binden an die α- und γ-Untereinheit. Jede Untereinheit ist ein Protein, das aus einem N-Terminus, vier transmembranären Segmenten (M1–M4) und einem C-Terminus besteht. Die M2-Segmente aller Untereinheiten bilden gemeinsam die Ionenkanal-Pore, durch die nach der Aktivierung des Rezeptors Chlorid-Ionen in das Neuron einströmen und die Reizweiterleitung erschweren.
Ähnliches gilt auch für die sogenannten Z‑Substanzen Zaleplon, Zolpidem und Zopiclon. Diese strukturell heterogenen, nicht von Benzodiazepinen abgeleiteten Arzneistoffe interagieren ebenfalls mit der α- und γ-Untereinheit des Rezeptors, allerdings bevorzugt bei solchen GABAA-Rezeptoren, die mit der α1-Isoform ausgestattet sind. (Benzodiazepine binden auch an die Isoformen 2, 3 und 5.) Auf diesem Unterschied beruht vermutlich die Beobachtung, dass Z‑Substanzen beruhigend und schlafanstoßend, aber kaum angst- und krampflösend wirken.
Positive allosterische Modulatoren an GABAA-Rezeptoren wie Benzodiazepine und Z‑Substanzen sind übrigens keine reine Erfindung des Menschen, sondern existieren auch im Organismus selbst. Eine Fülle von Arbeiten zeigen, dass bestimmte Neurosteroide (z.B. Metaboliten des Progesterons oder des Desoxycorticosterons) ebenfalls die Affinität von GABA zu dessen Bindungsdomänen erhöhen. Allerdings sind die Bindungsstellen nicht mit denen der Pharmaka identisch.
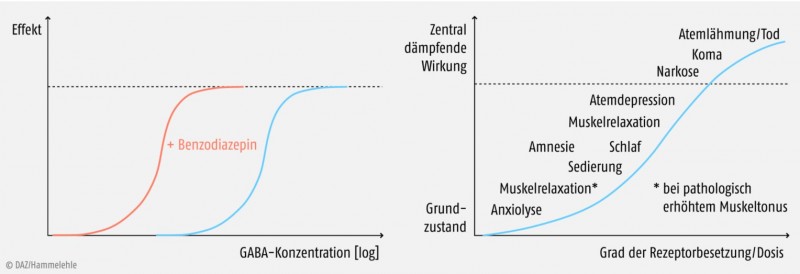
Die Tatsache, dass in Abwesenheit von GABA kein Effekt auftritt, unterscheidet Benzodiazepine und Z‑Substanzen ganz wesentlich von den Barbituraten. Diese aktivieren den GABAA-Rezeptor nämlich auch allein, sie sind also allosterische Agonisten. Ihre Maximaleffekte können sogar über die Wirkung von GABA hinausgehen (Abb. 2). Barbiturate wirken sedativ, hypnotisch und antikonvulsiv, aber nicht anxiolytisch. Ihr größter Nachteil besteht darin, dass sie bei höherer Dosierung narkotisch wirken, die autonome Regulation des Körpers stark beeinträchtigen und durch Lähmung der Atemmuskulatur den Tod hervorrufen können.
Benzodiazepin-Antagonist Flumazenil
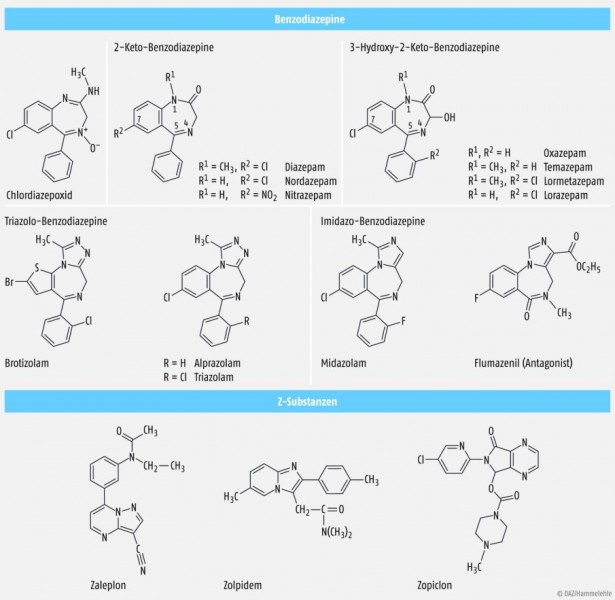
Das Imidazobenzodiazepin-Derivat Flumazenil (Abb. 3) ist streng genommen ein Partialagonist, der mit Benzodiazepinen um deren Bindungsstelle konkurriert. Über einen relativ großen Dosisbereich verdrängt es Benzodiazepine vom GABAA-Rezeptor, ohne eigene Effekte zu zeigen, und antagonisiert somit alle Benzodiazepin-Wirkungen. Daher wird es auch oft als Antagonist bezeichnet. Erst in sehr hoher Dosis wirkt Flumazenil selbst benzodiazepinähnlich. Es kann eingesetzt werden, um z.B. die hypnosedative Wirkung in der Allgemeinanästhesie aufzuheben oder auch um Symptome einer Benzodiazepin-Überdosierung zu bekämpfen. Zu beachten ist hierbei die relativ kurze Halbwertszeit von Flumazenil von ca. 60 min. Infusionen oder Injektionen müssen gegebenenfalls wiederholt werden bzw. der Patient muss bei einer einmaligen Gabe des Antidots über den Zeitraum des Abklingens hinaus engmaschig überwacht werden.
Struktur-Wirkungs-Beziehung, pharmakodynamische Eigenschaften
Für die Wirkung der Benzodiazepine sind folgende Strukturmerkmale wichtig: Ein bizyklisches System mit 7-gliedrigem Heterozyklus mit Stickstoffatomen an Position 1 und 4 (Diazepin), ein Phenylring an Position 5 und ein elektrophiler Substituent (z.B. Chlor, Brom oder Nitrogruppe) an Position 7 (Abb. 3). Eine Potenzierung der Wirkung kann durch Methylierung des Stickstoffs an Position 1 erreicht werden.
Heute auf dem Markt zu findende Benzodiazepine haben an Position 2 eine Ketogruppe (2-Keto- und 3-Hydroxy-2-keto-benzodiazepine) oder eine Aminogruppe (Chlordiazepoxid), oder sie haben einen fusionierten Imidazol- bzw. Triazolring (Abb. 3).
Betrachtet man die pharmakodynamischen Eigenschaften, lassen sich in Verdrängungsassays mit 3H-Flunitrazepam unterschiedlich starke Affinitäten zur Benzodiazepin- Bindungsstelle feststellen. Im Vergleich zu Chlordiazepoxid zeigen beispielsweise Lorazepam und Alprazolam eine sehr viel höhere Affinität, gefolgt von Diazepam, Bromazepam und Oxazepam. Clobazam und Dikaliumclorazepat haben eine relativ geringe Bindungsneigung. Es gilt: Je höher die Affinität, desto größer ist die sogenannte Potenz des Benzodiazepins, d.h. umso geringer ist die therapeutisch anzuwendende Dosis.
Derzeit ist es (noch) nicht möglich, durch strukturelle Abwandlungen eine gezielte Trennung der Benzodiazepin-Wirkungen zu erreichen. Bei einer anxiolytischen oder antikonvulsiven Therapie ist z.B. die Sedation eine oft unerwünschte Begleiterscheinung. Man sieht jedoch beim Eintreten der Effekte eine gewisse Dosisabhängigkeit. Ansteigende Dosierung ist in der Regel mit folgender Reihung korreliert, die einem zunehmenden Grad an Rezeptorbesetzung zugeschrieben wird: anxiolytisch – sedierend – schlafanstoßend – schlaferzwingend – zentral muskelrelaxierend (Abb. 2).Die drei GABAA-Rezeptor-Agonisten Zolpidem, Zaleplon und Zopiclon haben zwar keine Benzodiazepinstruktur, aber wie die Benzodiazepine ein stickstoffhaltiges, bizyklisches Strukturelement (Abb. 3). Zolpidem und Zaleplon besitzen eine gewisse Selektivität für die α1-Untereinheit des GABAA-Rezeptors, für Zopiclon ist dies nicht beschrieben. Die Bindungsaffinität der Z‑Substanzen ist im Vergleich zu Flunitrazepam deutlich schwächer.
GABAA-Rezeptoren und ihre Subtypen im ZNS
GABAA-Rezeptoren sind an synaptischen Schaltstellen im zentralen Nervensystem weit verbreitet. Sie kommen in großer Dichte in der Großhirnrinde und in mittlerer Dichte in Hypothalamus, Thalamus, Kleinhirn und limbischem System vor. In geringerem Ausmaß sind sie auch in Hirnstamm und Rückenmark zu finden. Da GABAerge Neuronen häufig als Interneuronen zwischengeschaltet sind, sind sie an nahezu allen Vorgängen im Gehirn beteiligt.
Benzodiazepine wirken hypnotisch (schlaffördernd), sedativ (beruhigend), anxiolytisch (angstlösend), antikonvulsiv (krampflösend) und muskelrelaxierend. Diese unterschiedlichen Effekte beruhen auf der bereits erwähnten Variabilität im Aufbau der GABAA-Rezeptoren. Mittels immunhistochemischer Untersuchungen und in Ligandenbindungsstudien konnte für einzelne Rezeptor-Subtypen eine Regioselektivität nachgewiesen werden:
- Regionen, die Schlaf und Wachheit regulieren (aufsteigende Bahnen des retikulären, thalamischen Systems), haben besonders viele GABAA-Rezeptoren mit α1-Untereinheit.
- Die angstlösende Eigenschaft der Benzodiazepine ist auf Rezeptoren mit α2-Untereinheit zurückzuführen (limbisches System).
- α2- und/oder α3-Untereinheiten finden sich vor allem in Bereichen, welche die zentrale Muskelrelaxation steuern (aus der Formatio reticularis absteigende Bahnen).
Ein weiterer Faktor, der die Wirkung der Benzodiazepine bestimmt, ist die lokale GABA-Konzentration in den jeweiligen Hirnarealen. Bei sehr hoher GABA-Konzentration im synaptischen Spalt kann durch allosterische Modulation nur noch eine vergleichsweise geringe Effektsteigerung erreicht werden. Dies bedeutet, dass Neuronen mit moderater Aktivierung ihrer GABAA-Rezeptoren am stärksten durch Benzodiazepine beeinflusst werden, wohingegen sehr aktive Neuronen in ihrer Aktivität kaum verändert werden.
Metabolisierung und Wirkdauer
Im Laufe der Zeit wurden zahlreiche neue Benzodiazepine synthetisiert, die zwar pharmakodynamisch dasselbe Target haben, sich jedoch in Wirkstärke und Pharmakokinetik voneinander unterscheiden.
Nahezu alle Benzodiazepine werden bei oraler Gabe gut aus dem Gastrointestinaltrakt resorbiert, sodass in der Regel Bioverfügbarkeiten von mehr als 80% erreicht werden. Die Substanzen besitzen eine hohe Plasmaeiweißbindung, je nach Lipophilie zwischen 70% (Alprazolam) und 99% (Diazepam).
Benzodiazepine werden hauptsächlich in der Leber metabolisiert. Hier finden in der Phase I Dealkylierungen (z.B. Diazepam zu Nordazepam), Hydroxylierungen (z.B. Nordazepam zu Oxazepam) und Reduktionen (z.B. der Nitrogruppe von Nitrazepam zur Aminoverbindung) statt. Anschließend werden die so veränderten Stoffe in Phase-II-Reaktionen meist mit Glucuronsäure konjugiert und als Glucuronide über den Urin ausgeschieden.
Aufgrund unterschiedlicher Metabolisierungswege und unter Berücksichtigung der Entstehung von aktiven Metaboliten kann man Benzodiazepine in Stoffe mit langer, mittlerer und kurzer Wirkdauer unterteilen (Tab. 1). Bei den langwirksamen Substanzen Diazepam und Chlordiazepoxid ist zu beachten, dass der Metabolit Nordazepam (syn. Desmethyldiazepam) eine noch längere Halbwertszeit von bis zu 90 Stunden hat. Dies kann bei mehrfacher Anwendung mit einem kurzen Dosierungsintervall insbesondere bei älteren Patienten schnell zu gefährlicher Kumulation führen.
Die Z‑Substanzen haben eine nur kurze Wirkdauer (Halbwertszeiten: Zaleplon etwa 1 h, Zolpidem 2 bis 3 h, Zopiclon 3 bis 6 h). Sie werden nach oraler Gabe gut resorbiert; die absolute Bioverfügbarkeit liegt für Zolpidem und Zopiclon zwischen 70 und 80%, für Zaleplon bei 30%. Zolpidem und Zaleplon werden in der Leber in inaktive Stoffwechselprodukte umgewandelt, welche renal und biliär eliminiert werden; Zopiclon und seine Metaboliten werden nahezu vollständig über die Niere ausgeschieden.
Anwendung als Hypnotika
Von einem idealen Schlafmittel ist zu fordern, dass es Einschlafen bzw. Durchschlafen fördert, keine unerwünschten Wirkungen wie Tagesmüdigkeit hervorruft, kein Abhängigkeitspotenzial bei längerer Anwendung besitzt, eine große therapeutische Breite aufweist und die Physiologie des natürlichen Schlafes nicht beeinflusst. Eine solche Substanz steht bis heute nicht zur Verfügung, daher sind der Anwendung von Benzodiazepinen hier bestimmte Grenzen gesetzt.
Benzodiazepine sollen nur angewendet werden, wenn sich die Schlafstörung durch nicht medikamentöse Maßnahmen (z.B. eine angepasste Schlafhygiene) nicht beseitigen lässt. Die Einnahmedauer sollte vier Wochen nicht überschreiten, um eine Toleranzentwicklung mit Dosissteigerungen zu vermeiden. Bei einer Einschlafstörung sind Benzodiazepine mit kurzer Halbwertszeit, wie Triazolam, Midazolam, oder die Z‑Substanzen Zolpidem und Zaleplon geeignet, bei Durchschlafstörungen werden häufig Temazepam, Lormetazepam, Nitrazepam oder Zopiclon (mittellange Halbwertszeit) eingesetzt.
Bei korrekter Anwendung über einen kurzen Zeitraum (2–4 Wochen) haben die Stoffe wenig Einfluss auf den wichtigen REM-Schlaf; die Schlafstadien II und III werden verlängert und das Stadium IV verkürzt. Nach Absetzen der Substanzen kann es zu einem unerwünschten REM-Rebound, d.h. einem erhöhten Anteil von REM-Schlafphasen, kommen. Daher sollte bei der Verordnung von Benzodiazepinen und Z‑Substanzen als Hypnotika die 5-K-Regel der Arzneimittelkommission der deutschen Ärzteschaft Beachtung finden:
1. Einsatz nur bei klarer Indikation.
2. Anwendung der kleinsten möglichen Dosis.
3. Anwendung über den kürzesten möglichen Zeitraum.
4. Kein abruptes Absetzen.
5. Kontraindikationen sind zu beachten.
Anwendung als Sedativa und Anxiolytika
Wegen ihrer sedativen und anxiolytischen Wirkung werden Benzodiazepine vor kleineren chirurgischen Eingriffen und zur Narkoseprämedikation eingesetzt. Hier sollen Angst und Stress des Patienten herabgesetzt werden. Einen großen Stellenwert haben die Substanzen heute auf der Intensivstation zur kontinuierlichen Sedierung des beatmeten Patienten, zumal bei Komplikationen sofort mit dem Benzodiazepin-Antagonisten Flumazenil eingegriffen werden kann.
Zur Vorbereitung von kleineren Eingriffen oder Gastroskopien ist Diazepam immer noch häufig in Gebrauch, da es in üblichen Dosen (5–10 mg oral) kaum unerwünschte Wirkungen auf die Atmung zeigt. Midazolam (0,15–0,2 mg/kg) wird wegen seiner geringen Nebenwirkungen und seiner kurzen Halbwertszeit oft zur Narkoseeinleitung eingesetzt.
Die anxiolytischen Effekte der Benzodiazepine nutzt man auch bei der Behandlung von Angst-, inneren Unruhe- und Spannungszuständen. Bei akuten psychischen Störungen mit Angst und Panikattacken sowie beim akuten Myokardinfarkt bilden sie eine wertvolle Behandlungsoption. Auch depressive Verstimmungen werden positiv beeinflusst. Der Einsatz bei Angsterkrankungen sollte jedoch immer von einer psychotherapeutischen Intervention begleitet werden.
Oft werden Benzodiazepine zur Überbrückung von besonders belastenden Lebenssituationen eingesetzt, wobei stets beachtet werden muss, dass sie den Umgang des Patienten mit seiner Situation zwar erleichtern können, jedoch keine Änderung der Situation an sich herbeiführen. Die Schwelle zum Missbrauch mit der Entwicklung einer Abhängigkeit ist hier oft schnell überschritten und eine qualifizierte Beratung durch den Apotheker von großem Nutzen.
Anwendung als Antikonvulsiva
Benzodiazepine setzen die zentrale Erregbarkeit herab und eignen sich daher auch zur Behandlung von Erkrankungen des epileptischen Formenkreises. In höherer Konzentration können sie die hochfrequenten Entladungen von Neuronen beim epileptischen Anfall unterdrücken. Jedoch eignen sie sich nur zur Übergangs- oder Krisenmedikation und nicht zur Dauertherapie, da ihre Wirkung aufgrund der Toleranzentwicklung immer schwächer wird. Ihre Hauptindikation ist der Status epilepticus. In dieser akuten Notfallsituation ist Diazepam (10–20 mg i.v.) das Mittel der ersten Wahl. Eine Ausnahme bildet Clonazepam, das bei Absence-Epilepsien und fokalen Epilepsien wirksam ist.
Anwendung als Muskelrelaxanzien
Benzodiazepine haben einen zentral muskelrelaxierenden Effekt. Hier kommt deren Angriff an der α2-Untereinheit des GABAA-Rezeptors im Rückenmark zum Tragen. Zentrale Muskelrelaxanzien sind indiziert bei schmerzhaften Spastiken der Skelettmuskulatur (z.B. durch zerebrale Läsionen) und bei pathologischen, schweren Muskelverspannungen. Hier wurde lange Zeit das Benzodiazepin Tetrazepam eingesetzt. Nachdem Anfang 2013 jedoch aufgrund des vermehrten Auftretens von schwerwiegenden Hautreaktionen die Nutzen-Risiko-Bewertung negativ ausfiel, ruht die Zulassung.
Unerwünschte Effekte und Suchtpotenzial
Die Einstufung einer Benzodiazepinwirkung als unerwünscht hängt entscheidend von der Indikation ab. Die am häufigsten auftretenden unerwünschten Wirkungen sind Sedation und Tagesmüdigkeit, Konzentrationsschwäche und verringerte Aufmerksamkeit mit einer verlängerten Reaktionszeit. Dadurch werden auch die Fähigkeit, Maschinen zu bedienen, und die Fahrtüchtigkeit beeinträchtigt. Eine Folge der zentralen Muskelrelaxation ist die Gangunsicherheit, die zu Stürzen führen kann. Ferner ist zu beachten, dass Benzodiazepine gerade bei älteren Patienten oft paradoxe Erregungszustände mit Reizbarkeit und Schlaflosigkeit auslösen. In dieser Patientengruppe ist das Risiko für Nebenwirkungen aufgrund des verlangsamten Abbaus der Substanzen generell erhöht. Benzodiazepine können zudem eine anterograde Amnesie auslösen.
Die dauerhafte Anwendung von Benzodiazepinen zur Bewältigung von Alltagssituationen ist immer als missbräuchlich zu betrachten. Die Patienten entwickeln eine Abhängigkeit: Sie können das Medikament nach längerer Anwendung nicht weglassen, weil dann Entzugssymptome (Schlaflosigkeit, Ängste, Schwindel, Dysphorie) auftreten. Werden sie hoch dosiert über einen längeren Zeitraum eingenommen, kann es beim Entzug sogar zu Depression, Panikattacken, Myalgien, Krämpfen und zum Delir kommen. Allerdings sind solche Fälle eher selten und in erster Linie bei Patienten mit multiplem Substanzmissbrauch zu finden.
Generell sind Benzodiazepine, korrekt eingesetzt, eine sichere Stoffgruppe. Selbst Dosen, welche in suizidaler Absicht eingenommen werden, führen in aller Regel nicht zum Tode, es sei denn, es werden noch andere zentral dämpfende Substanzen, z.B. Alkohol oder Opioide, beikonsumiert (vgl. Abb. 2). Benzodiazepine können die Plazentaschranke überwinden und auch in die Muttermilch übertreten. Dies kann beim Neugeborenen zu Sedierung und Muskelschwäche führen (sog. „floppy infant syndrome“).
Bei den Z‑Substanzen sollten die Nebenwirkungen aufgrund des selektiveren Angriffs an der α1-Untereinheit des GABAA-Rezeptors bezüglich der Muskelrelaxation (Sturzgefahr) und des Abhängigkeitspotenzials geringer ausfallen, weshalb sie für ältere Patienten geeigneter zu sein scheinen. Jedoch sind auch unter der Medikation mit Z‑Substanzen Stürze aufgetreten. Es besteht also auch hier ein gewisses Risiko, welches zu berücksichtigen ist. Bezüglich des Abhängigkeitsrisikos weisen die Z‑Substanzen nach den bisherigen Beobachtungen ebenfalls einen geringen Vorteil auf.
Aufsehen erregte eine im Jahr 2012 publizierte Studie, die bei älteren Patienten eine Assoziation (allerdings keinen kausalen Zusammenhang) zwischen der Einnahme von Benzodiazepinen und der Diagnose einer Demenz zeigte („Benzodiazepine use and risk of dementia“). Weitere Studien müssen hier folgen, um gesicherte Aussagen treffen zu können.
Absetzen von Benzodiazepinen nach Langzeitgabe
Hausärzte verordnen Benzodiazepine trotz der bekannten Bedenken relativ häufig über längere Zeit. Stattdessen sollten sie – wie auch die Apotheker – versuchen, dem Patienten auf behutsame Art und Weise ein Problembewusstsein für die negativen Effekte einer Dauereinnahme zu vermitteln. Hierbei ist es besonders wichtig, beim Patienten keine Abwehrhaltung zu erzeugen, sondern ihn zu motivieren, sich einer Entwöhnungstherapie zu unterziehen; hier ist die Erfolgsquote mit bis zu 90% außerordentlich hoch.
Grundsätzlich ist zu unterscheiden, ob es sich um eine Niedrigdosis-Abhängigkeit oder um eine Hochdosis-Abhängigkeit handelt. Letztere kommt häufig bei Patienten vor, die übermäßig Alkohol und/oder noch andere Drogen konsumieren. Hier sollte eine Entwöhnung aufgrund der zu erwartenden Entzugssymptome (z.B. Krampfanfälle) und einer möglichen Entzugspsychose stationär durchgeführt werden.
Bei der Niedrigdosis-Abhängigkeit haben die Patienten das Benzodiazepin über einen langen Zeitraum eingenommen, ohne die Dosis zu steigern, und können ambulant entwöhnt werden. Als Faustregel gilt, dass der Zeitraum der Entwöhnung in Monaten dem Zeitraum, über den das Medikament in Jahren eingenommen wurde, entspricht. Wurden vom Patienten Benzodiazepine mit relativ kurzer Halbwertszeit eingenommen, so ist es oft nicht möglich eine gleichmäßige, langsame Abnahme der Arzneistoffkonzentration im Körper zu erreichen. Deshalb kann es in diesen Fällen sinnvoll sein, zunächst schrittweise auf ein langwirksames Benzodiazepin wie Diazepam umzustellen und dann allmählich die Dosis zu reduzieren. Wenn der Patient an eine Tagesdosis gewöhnt war, die 120 mg Diazepam entspricht, kann die Entwöhnung in 40 Phasen von jeweils ein bis zwei Wochen erfolgen; insgesamt dauert sie also gut ein Jahr.
Patienten berichten, dass sie nach erfolgreicher Entwöhnung das Leben mit all seinen Facetten wieder sehr viel intensiver und realer erleben, und bewerten dies als sehr positiv. Daher ist es unabhängig davon, wie lange die Einnahme der Benzodiazepine angedauert hat oder wie alt der Patient ist, immer lohnenswert eine individuell abgestimmte Entwöhnung anzustreben.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.