













- DAZ.online
- DAZ / AZ
- DAZ 22/2013
- Arzneimittelzulassung in ...
AM-Zulassung
Arzneimittelzulassung in besonderen Fällen
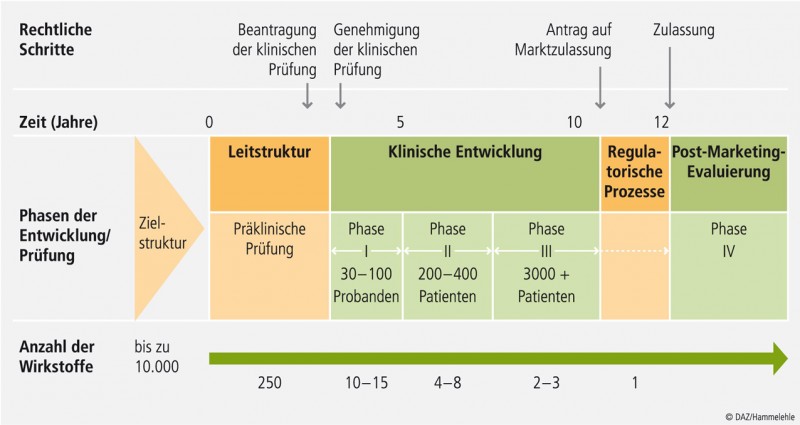
Abb. 1: Prozess von den Arzneistoffkandidaten bis zum zugelassenen Arzneimittel.
Bioäquivalenzstudien mit Generika
Für Generika, d. h. Arzneimittel mit einem Wirkstoff, der in einem bereits zugelassenen Arzneimittel enthalten ist, ist es relativ einfach, eine Zulassung zu bekommen. Bei ihnen ersetzt eine Bioäquivalenzstudie (BE-Studie; BE = bioequivalence) die pivotale Phase-III-Studie bei der Zulassung einer strukturell neuen Substanz. Eine BE-Studie testet nur an einer relativ geringen Anzahl von Probanden oder Patienten die Bioverfügbarkeit (area under the curve, AUC, das Integral der Plasmaspiegel-Zeit-Kurve) und den maximalen Plasmaspiegel (Cmax) des Generikums. Als bioäquivalent gilt hierbei eine Bioverfügbarkeit des Generikums von 80 bis 125% bezogen auf das Referenzprodukt, auf das im Zulassungsantrag Bezug genommen wird. Rechtliche Grundlage für dieses Procedere ist die aktuelle Fassung der Bioäquivalenz-Leitlinie von 1998 (BE-Guideline CHMP/EWP/QWP/1401/98 Rev. 1; CHMP = Committee for Medicinal Products for Human Use, Ausschuss für Humanarzneimittel der EMA; EWP = Efficacy Working Party; QWP = Quality Working Party; die aktuelle Version der BE-Guideline datiert vom 20. 01. 2010).
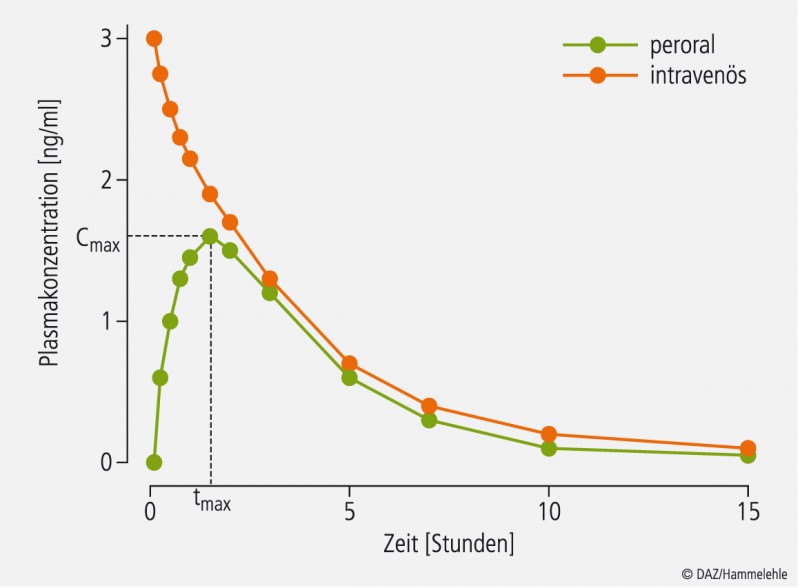
Abb. 2: Typische Plasmaspiegel-Zeit-Kurven von oral und intravenös applizierten Arzneimitteln. Alle i.v. applizierten Arzneimittel haben zum Zeitpunkt 0 eine Bioverfügbarkeit von 100%. Daher können Bioäquivalenzstudien entfallen.
Bei Substanzen mit einer engen therapeutischen Breite werden höhere Anforderungen an die Bioäquivalenz gestellt, während die Anforderungen bei Substanzen mit hoher intraindividueller Schwankungsbreite geringer sind (s. Textkasten "NTID und HVDP").
NTID und HVDPNarrow therapeutic index drugs (NTIDs) sind Substanzen mit einer engen therapeutischen Breite. Bei ihnen gelten strengere Maßstäbe für die Bioäquivalenz. Als bioäquivalent gilt hierbei eine Bioverfügbarkeit des Generikums von 90 bis 111% des Referenzprodukts (statt 80 – 125%). Deshalb muss bei NTIDs eine Bioäquivalenzstudie durchgeführt werden; eine Zulassung ohne klinische Daten wird in diesen Fällen nicht akzeptiert. Die Frage, ob ein Wirkstoff zu den NTIDs gehört, muss stets anhand des Einzelfalls diskutiert und entschieden werden. Die US-FDA hat eine Liste, in der alle NTIDs aufgeführt sind. In Europa gibt es keine solche Liste. Ein Beispiel für einen NTID ist der Wirkstoff Tacrolimus (Originator: Prograf®). Als highly variable drug products (HVDPs) definiert die BE-Guideline solche Substanzen, deren Wirksamkeit eine intraindividuelle Variabilität von über 30% besitzen. Bei HVDPs kann das Akzeptanzintervall vergrößert werden, und zwar maximal auf 70 bis 143% (bei einer intraindividuellen Variabilität über 50%). Ein Beispiel für ein HVDP ist der Wirkstoff Clopidogrel (Originator: Plavix®). |
Nicht so weitläufig bekannt sind die (relativ vielfältigen) Möglichkeiten, eine generische Marktzulassung ohne die Erhebung klinischer Daten zu erlangen. Im Folgenden werden die vier wichtigsten Möglichkeiten dargestellt (Überblick in Tab. 1).
Tab. 1: Rechtliche Grundlagen gemäß Richtlinie (Directive) 2001/83/EC, um auf europäischer Ebene die Zulassung für Generika zu beantragen | |
Quelle |
Zulassungsart |
|
Artikel 10
Abs. 1
|
Bezugnehmende Zulassung mit einer BE-Studie, die die pivotale Phase III des Originators ersetzt. |
|
Artikel 10
Abs. 2b
|
Bezugnehmende Zulassung, bei der ein Biowaiver (Verzicht auf die Durchführung eine BE-Studie) gewährt werden kann, wenn die Kriterien der BE-Guideline erfüllt sind. |
Artikel 10a |
Der sogenannte bibliografische Antrag kommt zum Tragen, wenn das Originator-Produkt mindestens 10 Jahre im Markt ist (Well-established use, WEU). In diesem Fall kann sowohl der präklinische als auch der klinische Teil des Dossiers durch adäquate wissenschaftliche Literatur ersetzt werden. |
|
Artikel 10
Abs. 3
|
Sogenannte Hybrid-Anträge: In Fällen, in denen es nicht sinnvoll ist, die Bioäquivalenz anhand der Plasmaspiegel nachzuweisen, kann es ausreichend sein, die therapeutische Äquivalenz anhand von Surrogat-Parametern nachzuweisen.
Bsp.: Im Fall der Gonadorelin-Superagonisten zur Therapie des Prostatakarzinoms ist es wichtiger, die Suppression des Testosterons (Surrogat) nachzuweisen als die Arzneistoffkonzentration. (Testosteron ist der Wachstumstreiber des Tumors)
|
Die Standardzulassung
Die Standardzulassung gemäß § 67 Abs. 5 AMG stellt bestimmte Arzneimittel von der Zulassungspflicht frei. Dabei handelt es sich im Regelfall um lang bekannte und gut untersuchte Arzneimittel oder um pflanzliche Arzneimittel. Nach AMG ist die Voraussetzung für eine Standardzulassung, dass eine Gefährdung der Gesundheit nicht zu befürchten ist, weil die Anforderungen an Qualität, Wirksamkeit und Unbedenklichkeit erwiesen sind. Beispiele für Standardzulassungen sind Paracetamol und Acetylsalicylsäure (ASS). Eine Liste der Standardzulassungen findet man unter: www.bfarm.de/DE/Arzneimittel/2_zulassung/verfahren/stdZul/stdzul-node.html.
Generika zur intravenösen Applikation
Ein wichtiges Charakteristikum für Generika ist ihre Austauschbarkeit mit dem Originalpräparat, auf das sich ihre Zulassung bezieht. Daher bezeichnet man Generika-Zulassungen, die sich an einen Originator anlehnen, auch als bezugnehmende Zulassungen. Dieser Umstand bedingt, dass das Generikum mit dem Originator austauschbar ist (cave: ein Generikum ist nur mit dem Originator austauschbar, nicht mit anderen Generika, auch dann nicht, wenn sie auf den gleichen Originator Bezug nehmen). Dies spiegelt sich in einer allgemeinen Definition eines Generikums wider: "Als Generika gelten Arzneimittel, die sich bezüglich ihres Wirkstoffes, ihrer Darreichungsform und ihrer Dosierung an ein zugelassenes Originalpräparat anlehnen. Sie sind mit den Originalpräparaten austauschbar."
Die Austauschbarkeit wird mittels einer BE-Studie belegt, wobei üblicherweise in einem Cross-over-Design die Probanden beider Studienarme einmal den Originator und einmal das Generikum (reference product bzw. test product) erhalten.
Einen Sonderfall stellen intravenös zu applizierende Arzneimittel dar, weil sie direkt nach der Applikation zu 100% bioverfügbar sind. Eine Bioverfügbarkeitsstudie wäre bei ihnen sinnlos und muss vom Antragsteller nicht vorgelegt werden.
Bibliografischer Zulassungsantrag
Wenn ein Arzneimittel bereits länger auf dem Markt ist und neben den Zulassungsunterlagen weiteres wissenschaftliches Erkenntnismaterial vorliegt, kann der Generikahersteller auch darauf Bezug nehmen. So sind beispielsweise einige Glucocorticoide und andere Endokrinologika bereits in den 1950er oder 1960er Jahren in den Markt eingeführt worden, über die es hunderte wenn nicht sogar tausende wissenschaftliche Publikationen, klinische Studien und auch Sekundärliteratur (Metaanalysen, systematische Reviews) gibt. Da solche Substanzen bereits viele Tausend Patientenjahre an Erfahrung kumulativ verbuchen können, werden für die Zulassung von Generika oftmals keine klinischen Daten mehr gefordert. Dies gilt allerdings nur, wenn Wirkstoff, Hilfsstoffe und Darreichungsform identisch sind mit bereits im Markt befindlichen Präparaten. Anderenfalls läuft der Antragsteller Gefahr, dass die Zulassungsbehörde von ihm klinische Daten fordert.
BCS-basierte Biowaiver
Ein Biowaiver ist die behördliche Zustimmung zum Verzicht auf die Durchführung einer Bioäquivalenzstudie (Vergleich Generikum mit Originator). Das biopharmazeutische Klassifizierungssystem (BCS) teilt Arzneistoffe aufgrund ihrer Löslichkeit in wässrigen Lösungen und ihrer intestinalen Permeabilität ein (Tab. 2). Ein BCS-basierter Biowaiver kann für schnell freisetzende orale Darreichungsformen, also einen leicht löslichen Wirkstoff (BCS-Klasse I oder III), beantragt werden. Bei BCS-Klasse I müssen in vitro mindestens 85% des Wirkstoffs innerhalb von 30 Minuten freigesetzt werden und bei BCS-Klasse III mindestens 85% innerhalb von 15 Minuten. Hilfsstoffe dürfen die (intestinale) Bioverfügbarkeit nicht beeinflussen (BE-Guideline, Appendix III).
Tab. 2: Das biopharmazeutische Klassifizierungssystem (BCS) mit Beispielen | ||
Klasse |
Eigenschaften des Arzneistoffs |
Beispiele |
I |
Löslichkeit↑ Permeabilität↑ |
Metoprolol, Diltiazem, Verapamil |
II |
Löslichkeit↓ Permeabilität↑ |
Diclofenac, Glibenclamid, Ibuprofen |
III |
Löslichkeit↑ Permeabilität↓ |
Cimetidin, Ranitidin, Aciclovir |
IV |
Löslichkeit↓ Permeabilität↓ |
Taxole, Furosemid, Hydrochlorothiazid |
Aktuelle Entwicklung im europäischen Kontext
Die Frage, ob ein Biowaiver gewährt werden kann für ein bestimmtes Produkt, wird auf nationaler wie europäischer Ebene gelegentlich auch kontrovers diskutiert. Da die Erhebung klinischer Daten kostspielig ist, hat die pharmazeutische Industrie ein Interesse an Biowaivern. Während sich die BE-Guideline inhaltlich recht eindeutig in dieser Fragestellung positioniert, ist die Frage, wie viel Ermessensspielraum sie Antragstellern und Behörden lässt, weniger klar und hat in der Vergangenheit zu Referral-Verfahren geführt (s. Textkasten "Arten von Referrals").
Arten von ReferralsEin Referral ist eine Rückgabe bei Nichteinigung im Zulassungsverfahren an das CHMP. Es gibt verschiedene Gründe für ein Referral: Sie reichen von Sicherheitsbedenken bis zu Meinungsverschiedenheiten über die Anwendungsgebiete des Arzneimittels. Referrals können von der Europäischen Kommission, einem Mitgliedstaat oder vom pharmazeutischen Unternehmer beantragt werden. Rechtliche Grundlage ist die Richtlinie (Directive) 2001/83/EC (siehe Tab. 3 zu den unterschiedlichen Verfahren). Abgeschlossene Referrals können eingesehen werden auf der Homepage der Europäischen Kommission: http://ec.europa.eu/health/documents/ community-register/html/index_en.htm |
Im Jahr 2011 wurde beispielsweise ein Referral-Verfahren gemäß Artikel 29 (Tab. 3) auf der Homepage der Europäischen Kommission publiziert, in dem das CHMP einen Biowaiver für eine orale Lösung des Wirkstoffs Dexamethason befürwortet. Dies geschah auf der wissenschaftlichen Basis, dass Dexamethason rasch löslich und bezüglich Biomembranen gut permeabel ist und daher eine hohe Bioverfügbarkeit besitzt. Allerdings wies das CHMP im Begründungstext des Referral-Verfahrens auch darauf hin, dass diese Entscheidung der BE-Guideline widerspricht (EMEA/H/A-29/1308). Denn eigentlich sind BCS-basierte Biowaiver gleichen Darreichungsformen vorbehalten, und eine Übertragung von einer Tablette auf eine orale Lösung ist nicht vorgesehen. In der Folge stellt sich die Frage, ob dieses Referral ein Präzedenzfall ist, ob es nur für die Substanzklasse der Glucocorticoide ein Präzedenzfall ist oder ob es eine Einzelfallentscheidung bleibt. Gegenstand der gegenwärtigen Entscheidungsfindung auf europäischer Ebene ist demzufolge auch, wie man in den Mitgliedstaaten der EU Inkonsistenzen in der Entscheidungsfindung zu Biowaivern vermeidet.
Tab. 3: Die unterschiedlichen Referral-Verfahren gemäß Richtlinie (Directive) 2001/83/EC | ||
Quelle |
Inhalt |
Anwendung |
Artikel 13 |
Harmonisation, Verfahren der gegenseitigen Anerkennung und Dezentrales Verfahren (s. u.: Artikel 29 Abs. 4) |
Bei Meinungsverschiedenheiten zwischen den Mitgliedstaaten über eine Variation (Typ II) |
Artikel 20 |
Sicherheits-, Qualitäts-, Herstellungs-, oder Wirksamkeitsbedenken
(safety, quality, manufacturing or
efficacy issues) |
Bei Arzneimitteln, die durch das zentralisierte Verfahren zugelassen wurden, hinsichtlich Sicherheits- und Herstellungsbedenken |
Artikel 29 |
Arzneimittel für Kinder
(paediatric medicine issues)
|
Wenn eine neue Indikation oder eine neue Darreichungsform für die Anwendung bei Kindern besteht |
|
Artikel 29
Abs. 4
|
Harmonisation, Verfahren der
gegenseitigen Anerkennung und Dezentrales Verfahren
(harmonisation,
mutual-recognition procedure = MRP, decentralised procedure = DPC) |
Wenn Unstimmigkeit zwischen den Mitgliedstaaten
bezüglich eines Arzneimittels während eines DPC- oder MRP-Verfahrens bestehen oder wenn eine potenzielle schwerwiegende Gefahr für die öffentliche Gesundheit besteht
|
Artikel 30 |
Wenn die Mitgliedstaaten unterschiedliche Entscheidungen für ein Arzneimittel (z. B. verschiedene Indikationen, Kontraindikationen oder Dosierung) gefällt haben oder wenn es nötig ist, innerhalb der EU zu harmonisieren |
|
Artikel 31 |
Sicherheits-, Qualitäts-, Herstellungs-,
oder Wirksamkeitsbedenken (s. o.: Artikel 20)
|
Wenn Bedenken bezüglich der Qualität, Sicherheit oder Wirksamkeit eines Arzneimittels oder einer Klasse von Medikamenten bestehen |
Artikel 107 i |
Sicherheitsbedenken
(safety issues)
|
Wenn ein Mitgliedstaat oder die Europäische Kommission der Auffassung ist, dass Sicherheitsbedenken bestehen. Dieses Referral umfasst: Widerruf der Zulassung oder größere Änderungen an der Zulassung wie das Löschen von Indikationen, die Verringerung der empfohlenen Dosis oder neue Kontraindikationen |
Extrapolation klinischer Daten
Eine andere Frage lautet, ob man klinische Daten bezüglich der Wirkstärke extrapolieren darf. Anders formuliert: Wenn man für die Zulassung eines Generikums klinische Daten erheben (und teuer bezahlen) muss, darf man dann in Bioäquivalenzstudien nur eine Wirkstärke testen und auf andere Wirkstärken extrapolieren, für die man ebenfalls eine Zulassung erhalten möchte?
Generell lautet die Antwort: Ja, es gibt Situationen, die eine Daten-Extrapolation auf andere Wirkstärken erlauben. Mit der Bioäquivalenz-Leitlinie konform ist es typischerweise, wenn man Bioäquivalenz für den höchsten Wirkstoffgehalt (z. B. einer Tablette) mit klinischen Daten nachweist und die geringeren Wirkstärken extrapoliert, wenn der Arzneistoff eine lineare Pharmakokinetik besitzt. Ein solches Vorgehen spart dem Unternehmer viel Geld.
Zulassung von innovativen Substanzen bei unvollständigen klinischen Daten
Anders ist die Situation bei Neuzulassungen im Bereich innovativer Arzneistoffe. Hier wird die zuständige Behörde in jedem Fall klinische Daten verlangen. Doch welche Möglichkeiten gibt es, wenn die betroffenen Patienten rar oder nur schwer zu erreichen sind? Der Gesetzgeber sieht hierfür auf europäischer Ebene zwei regulatorische Instrumente vor:
1. die (ältere) Form der Zulassung unter besonderen Umständen (Marketing Authorisation under exceptional circumstances)
2. die (neuere) Form der Zulassung mit Auflage (Conditional Marketing Authorisation)
Beide Antragsformen werden in Tabelle 4 einander vergleichend gegenübergestellt.
Tab. 4: Vergleich von zwei möglichen Zulassungsverfahren bei innovativen Substanzen (MA = Marketing Authorisation, Marktzulassung) | |
Conditional MA* |
MA under exceptional circumstances** |
|
Wird erteilt, bevor alle Daten verfügbar sind, um dringend benötigte Arzneimittel schnell bereitzustellen:
Kriterien und Anforderungen:
Die zunächst auf vorläufigen Erkenntnissen statt auf umfangreicher Entwicklung basierende Genehmigung ist für ein Jahr gültig und verlängerbar
Nach Erfüllung der Auflagen kann eine "normale" Zulassung gewährt werden
|
Vollständige Einreichung der Unterlagen nicht möglich wegen/weil:
Kriterien und Anforderungen:
Die Aufrechterhaltung der Genehmigung ist von der jährlichen Neubeurteilung dieser Bedingungen abhängig
In der Regel folgt keine "normale" Zulassung
|
| * Regulation (EC) No. 726/2004, Titel II, Kapitel 1 Art. 14 Abs. 7 ** Regulation (EC) No. 726/2004, Titel II, Kapitel 1 Art. 14 Abs. 8 | |
Fazit
Im Generikabereich gibt es verschiedene Arten der Arzneimittelzulassung ohne die Vorlage klinischer Daten, wenn bestimmte Voraussetzungen vorliegen. Die wichtigsten Arten sind die Standardzulassung nach AMG, der bibliografische Zulassungsantrag (well-established use application) nach Directive 2001/83/EC und die Beantragung eines BCS-basierten Biowaivers nach den Kriterien der Bioäquivalenz-Leitlinie. Die beiden Letztgenannten geben gelegentlich Anlass zu einer tiefergehenden wissenschaftlichen Erörterung, die im europäischen Kontext als Referral-Verfahren durchgeführt werden kann.
Bei strukturell neuen Substanzen kann der Hersteller nur in besonderen Ausnahmefällen eine Zulassung erreichen, obwohl die klinische Datenlage unzureichend ist. Wenn ein besonderes Interesse der Patienten oder der Öffentlichkeit vorliegt, reicht eine (vorläufig) positive Nutzen-Risiko-Abwägung aus.
Weiterführende Literatur Eckstein N, Haas B. Vom Wirkstoff zum Arzneimittel, 2. Teil: Klinische Prüfungen. Dtsch Apoth Ztg 2012;152(29):61 – 67. Gothoskar AV. Biopharmaceutical classification of drugs. Pharm Rev [Online] 2005, 3 (1). www.pharmainfo.net/reviews/biopharmaceutical-classification-drugs. Heinzl S. Klinische Studien: Wie Arzneimittel geprüft werden. Pharm Ztg 2011;156(30):16 – 20. Guideline on the scientific application and the practical arrangements necessary to implement commission regulation (EC) No 507/2006 on the conditional marketing authorization for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004. Schwarz JA. Leitfaden klinische Prüfungen von Arzneimitteln und Medizinprodukten. 4. Aufl. Aulendorf 2011.
Autoren
Dr. Niels Eckstein ist Apotheker, promovierter Pharmakologe und Lehrbeauftragter der FH Köln für Pharmamanagement. Kontakt: Niels.Eckstein@web.de
Dr. Bodo Haas ist Apotheker, promovierter Pharmakologe und Lehrbeauftragter der FH Köln für Pharmakokinetik.
Lea Röper studiert pharmazeutische Chemie im Schwerpunktfach Pharmamanagement an der FH Köln.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.