



















- DAZ.online
- DAZ / AZ
- DAZ 6/2012
- Stereochemie
UniDAZ
Stereochemie
Was Pharmaziestudierende darüber wissen sollten
Liebe Leserinnen, liebe Leser,die Rubrik UniDAZ richtet sich insbesondere, aber keineswegs ausschließlich, an den pharmazeutischen Nachwuchs, also an Studierende, Pharmazeuten im Praktikum und junge Apothekerinnen und Apotheker. UniDAZ will pharmazeutisches Grundlagenwissen vermitteln bzw. auffrischen oder an Beispielen die Anwendung pharmazeutischen Wissens in der Forschung zeigen. Dabei hoffen wir, dass diese Beiträge für Sie nicht nur fachlich interessant sind, sondern Ihnen auch bei der Examensvorbereitung nützen. Nicht zuletzt deshalb widmet die Rubrik UniDAZ jedem Prüfungsfach des zweiten Staatsexamens einen Beitrag pro Semester. Der Beitrag "Flotierende Arzneiformen" behandelt ein Thema der Pharmazeutischen Technologie und ist bereits der vierte Beitrag der Rubrik UniDAZ. Von Oktober bis Dezember 2011 erschienen:
Zur "Marke" UniDAZ gehören auch das Magazin, das zu Beginn jedes Semesters an den pharmazeutischen Instituten verteilt wird, und die Website www.unidaz.de, auf der Sie die Beiträge des Magazins nachlesen und kommentieren können. Bei den Hochschulporträts und den Blogs können Sie sogar selbst als Autor tätig werden. Wir wünschen Ihnen viel Vergnügen beim Lesen und eventuell auch beim Schreiben. Ihre UniDAZ-Redaktion |
Chiralität
Der Begriff Chiralität, der von griech. χειρ (cheir) = Hand abgeleitet ist und den man auch Händigkeit nennen kann, bezeichnet die Eigenschaft eines Objektes, sich von seinem Spiegelbild zu unterscheiden. Ein Molekül, oder allgemeiner formuliert, ein Objekt ist dann chiral, wenn es durch Symmetrieoperationen wie Rotation, Parallelverschiebung, Umkehrung, Flächenspiegelung oder Drehspiegelung mit seinem Spiegelbild nicht zur Deckung gebracht werden kann. Ein chirales Objekt verhält sich zu seinem Spiegelbild wie die rechte Hand zur linken Hand.
Zu unterscheiden sind zentrale, axiale und planare Chiralität. Obwohl bei den Arzneistoffen das asymmetrisch substituierte C-Atom die wichtigste und häufigste Voraussetzung für das Vorliegen chiraler Moleküle ist, sind alle anderen Moleküle, die keine Symmetrieebene aufweisen, ebenso chiral.
Spiegelbildliche Paare chiraler Verbindungen nennt man Enantiomere, abgeleitet von griech. εναντιος (enantios) = entgegengesetzt. Architekten und Biologen kennen Enantiomorphe als Paare spiegelbildlicher Objekte, wie beispielsweise rechts- und linkswindige Säulen oder rechts- und linksgängige Schneckenhäuser.
Symmetrie, Asymmetrie und optische Aktivität
Ein Objekt (auch ein Molekül) ist symmetrisch, wenn man es durch räumliche Umorientierung in einen äquivalenten, also von der Ausgangslage nicht unterscheidbaren Zustand überführen kann; wenn das nicht möglich ist, ist es asymmetrisch. Die in Tabelle 1 zusammengefassten Symmetrieoperationen (Art der Umorientierung) und Symmetrieelemente (zugehörige Operatoren) sind ohne Einschränkung auf Moleküle anwendbar, weil diese dreidimensionale Objekte darstellen.
Die gesamten Symmetrieoperationen, die an einem Molekül durchführbar sind, nennt man Punktgruppen. Sie werden durch die Schönflies-Symbole gekennzeichnet, die sich auf die Symmetrieelemente beziehen und deren Indices (n) die Anzahl der möglichen Symmetrieoperationen nennen, nach deren Ausführung der Ausgangszustand wieder erreicht ist (Tabelle 1 u. Tabelle 2).
tab. 1: Grundbegriffe der Stereochemie |
||||
| Symmetrieoperation | Schönflies- Symbol | Symmetrieelement | Chira- lität | Optische Aktivität |
| Spiegelung (Reflexion, Flächenspiegelung) | σ (oder s) | Spiegelebene, Symmetrieebene | achiral | — |
| Drehung (Rotation) | Cn (n = 1, 2, 3 …) |
Drehachse, Symmetrieachse | achiral | — |
| Umkehrung (Inversion, Punktspiegelung) | i | Inversionszentrum, Symmetriezentrum | chiral | + |
| Drehspiegelung (Drehung + Spiegelung) | Sn (n = 1, 2, 3 …) |
Drehspiegelachse | chiral | + |
| tab. 2: Schönflies-Symbole (vgl. Tab. 1) | |
| Cn | (von cyclisch) Gruppe mit einer n-zähligen Symmetrieachse |
| S, Sn | (von Spiegelung) Gruppe ohne Symmetrieelement bzw. mit einer n-zähligen Drehspiegelachse |
| Dn | (von Dieder, sprich: Di-eder) diedrische Symmetrie, Gruppe mit einer n-zähligen Hauptachse und n zweizähligen Nebenachsen |
| t | (von tetraeder) tetraedrische Systeme, Gruppe mit vier drei- zähligen und sechs zweizähligen Achsen (in Würfelform) |
| O | (von Oktaeder) oktaedrische Symmetrie, Gruppe mit drei vier- zähligen und sechs zweizähligen Achsen (in Würfelform) |
| K | sphärische Symmetrie, Gruppe mit allen Symmetrieelementen |
| d, h, v | diagonale, horizontale, vertikale Symmetrieebene |
Von symmetrisch bis asymmetrisch
Symmetrisch heißt spiegelbildlich übereinstimmend (s. o.). Symmetrieverwandt sind Objekte oder Strukturformeln, wenn sie den gleichen Satz von Symmetrieelementen besitzen.
Dissymmetrisch sind Moleküle mit Drehachsen (Cn), aber ohne Symmetrieebenen, Symmetriezentren oder Drehspiegelachsen. Im heutigen Sprachgebrauch werden dissymmetrisch und chiral synonym verwendet.
Asymmetrisch sind Moleküle, wenn sie kein Symmetrieelement aufweisen. Dissymmetrische Moleküle brauchen nicht asymmetrisch zu sein, um Chiralität zu besitzen, doch sind alle asymmetrischen Moleküle zwangsläufig dissymmetrisch.
Voraussetzung für die optische Aktivität ist die Chiralität (Tab. 1).
Symmetrieelemente
Ein Molekül verfügt über eine Symmetrieachse (Cn), wenn es durch axiale Drehung um einen bestimmten Winkel mit seiner Ausgangslage zur Deckung gebracht werden kann.
Eine Symmetrieebene (σ) teilt ein Molekül in der Weise, dass jede Hälfte das Spiegelbild der anderen Hälfte darstellt.
Ein Symmetriezentrum (Inversionszentrum, i) enthält ein Molekül, wenn jedes seiner Atome durch zentrale Spiegelung mit einem korrespondierenden Atom zur Deckung gebracht werden kann.
Über eine Drehspiegelachse verfügt ein Molekül, wenn es durch Drehung und Spiegelung wieder in seine Ausgangslage zurückfindet.
Optische Aktivität
Die Fähigkeit eines Stoffes, die Schwingungsebene von linear polarisiertem Licht zu drehen, wird als optische Aktivität bezeichnet. Bei organischen Verbindungen ist die Chiralität Voraussetzung für diese Fähigkeit, die für jedes Molekül eine charakteristische Größe aufweist.
Enantiomere, die sich in ihren physikalischen Eigenschaften nicht unterscheiden, differieren allerdings in ihrer optischen Aktivität. Unter gleichen Messbedingungen drehen sie die Ebene des linear polarisierten Lichtes um den gleichen Betrag in entgegengesetzter Richtung. Man nennt Enantiomere deshalb "optische Antipoden". Als (+)-Enantiomer wird die nach rechts drehende und als (–)-Enantiomer die nach links drehende Verbindung bezeichnet. Die äquivalente Mischung beider Enantiomere zeigt wegen der Aufhebung der entgegengesetzten Drehwerte keine optische Aktivität; sie wird als Racemat bezeichnet.
Enantiomere in achiraler und in chiraler Umgebung
Befinden sich Enantiomere in einem achiralen Milieu, beispielsweise in wässriger, ethanolischer oder acetonischer Lösung, so verhalten sie sich – abgesehen von ihrer entgegengesetzten optischen Aktivität – in chemischer und physikalischer Hinsicht völlig gleichartig. In chiraler Umgebung oder mit chiralen Agenzien reagieren sie verschiedenartig und verschieden schnell. Durch dieses Verhalten wird die Auftrennung von Racematen in Enantiomere mithilfe chiraler Reagenzien ermöglicht und lässt sich die unterschiedliche biologische, pharmakologische und sensorische Wirkung erklären. Da die Rezeptoren im menschlichen Körper und alle Enzyme aus dem Bereich der gesamten Biologie aus chiralen Molekülen bestehen oder aufgebaut sind, ist ihre unterschiedliche Affinität und Reaktivität gegenüber Enantiomeren nicht nur erklärbar, sondern sogar zu erwarten. Gegenüber chiralen Rezeptoren und Enzymen verhalten sich Enantiomere wie zwei unterschiedliche Verbindungen.
Zentrale Chiralität
Wenn es um chirale Arzneistoffe geht, dann nehmen solche mit einem "asymmetrischen C-Atom" – korrekt formuliert: mit einem asymmetrisch substituierten C-Atom – den breitesten Raum ein.
Zur Kennzeichnung der Konfiguration existieren zwei Konventionen, die traditionelle Fischer-Konvention und die rationale Cahn-Ingold-Prelog-Konvention.
Fischer-Konvention
Die Symbole d (von dexter = rechts) und l (von laevus = links) wurden von Emil Fischer für die stereochemische Kennzeichnung von Zuckern eingeführt und beziehen sich auf das Asymmetriezentrum des Glycerinaldehyds als definitionsgemäß einem der einfachsten Zucker (Aldotriose). Der andere ist das achirale Dihydroxyaceton (Ketotriose).
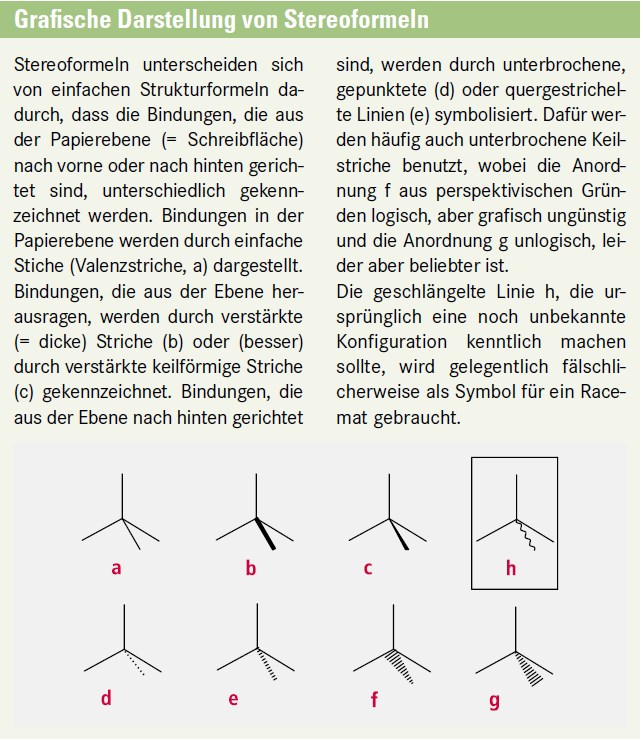
Die Fischer-Projektion projiziert den dreidimensionalen Kohlenstoff-Tetraeder in die zweidimensionale Ebene. Dabei sind folgende Punkte zu beachten:
Die längste Kohlenstoff-Kette ist vertikal anzuordnen.
Das höher oxidierte Ende der Kette steht oben.
Die horizontalen Bindungen weisen aus der Betrachtungsebene nach vorne.
Die vertikalen Bindungen an jedem C-Atom der Hauptketten weisen nach hinten.
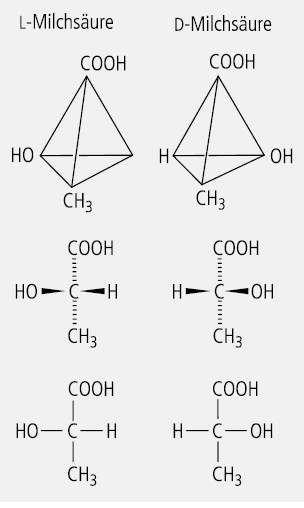
In Abbildung 1 ist am Beispiel der Milchsäure dargestellt, wie man, von der Außenansicht eines entsprechend substituierten Tetraeders ausgehend, die stereochemischen Bindungsrichtungen durch Keilstriche und gestrichelte Linien ableitet und diese dann durch einfache Valenzstriche ersetzt.
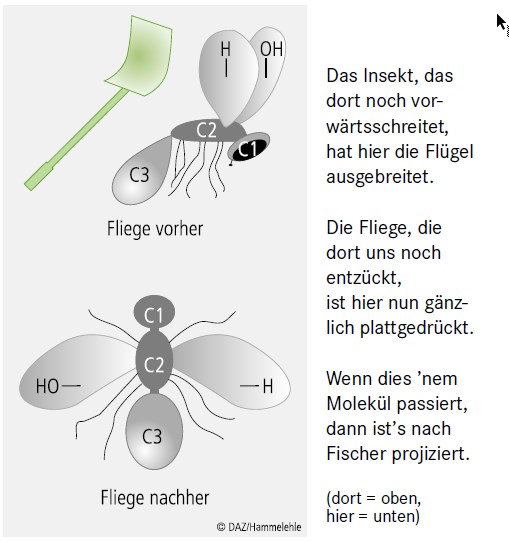
Der Betrachter, der diese Regeln kennt, kann von der zweidimensionalen Fischer-Projektion auf die räumliche Struktur des Moleküls zurückschließen. In Abbildung 2 ist der Prozess der Fischer-Projektion mit einem Begleittext allgemeinverständlich dargestellt (nach Prof. Dr. Armin Wolff, Hochschule Albstadt-Sigmaringen).
Zeigt die funktionelle Gruppe (OH) nach rechts, so liegt eine d-Form vor, zeigt sie nach links, dann handelt es sich um eine l-Form. Die Symbole d und l geben die konfigurative Beziehung zum d- und l-Glycerinaldehyd an, stellen aber keinen Bezug zur optischen Drehrichtung her. Diese physikalische Eigenschaft wird durch ein (+) für rechtsdrehend und ein (–) für linksdrehend gekennzeichnet.
Das grundlegende Experiment Emil Fischers, das zu einer konsequenten stereochemischen Bezeichnungsweise geführt hat, war der Abbau der (+)-Glucose zum D(+)-Glycerinaldehyd.
Die Verwendung des Glycerinaldehyds (Abb. 3) als Bezugssystem für die konfigurative Zuordnung der Kohlenhydrate hat sich durchgesetzt und ist heute noch sinnvoll. Bei anderen Verbindungsklassen, beispielsweise den Aminosäuren, führt sie aber zu widersprüchlichen Ergebnissen. Die d- und l-Nomenklatur gibt lediglich die relative Konfiguration an.
Cahn-Ingold-Prelog-Konvention (CIP)
Zur Beschreibung der absoluten Konfiguration haben die Chemiker R. S. Cahn, C. Ingold und V. Prelog 1951 die nach ihnen benannte CIP-Konvention geschaffen. Anstelle von d und l verwendet die CIP-Nomenklatur R (von lat. rectus = rechts) und S (von lat. sinister = links).
Die Zuordnung an einem Chiralitätszentrum (Stereozentrum) eines Moleküls verläuft nach einer Prioritätsfolge. Ein Stereozentrum eines Arzneistoffs besteht meistens aus einem C-Atom, das vier verschiedene Substituenten trägt. Bei anderen Atomen wie Stickstoff in einer starren Fixierung (Beispiel China-Alkaloide) oder Schwefel (Beispiel Omeprazol) kann auch ein freies Elektronenpaar die Rolle eines Substituenten übernehmen.
Direkt am C-Atom gebundene Substituenten mit Heteroatomen haben Vorrang vor solchen mit C-Atomen. Es gilt die Prioritätsfolge:
I > Br > Cl > S > P > F > O > N > C > H
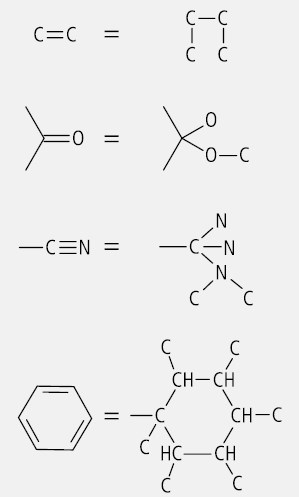
Das Atom mit der höchsten Ordnungszahl im Periodensystem erhält die Ziffer 1, das mit der zweithöchsten Ordnungszahl die Ziffer 2, das mit der geringeren die Ziffer 3 und das Element mit der niedrigsten Ordnungszahl (fast immer der Wasserstoff) die Ziffer 4. Eine noch niedrigere Priorität haben freie Elektronenpaare. Anstelle der Ziffern können auch die Buchstaben a bis d verwendet werden. Bei Substituenten mit gleicher Ordnungszahl hat jener Priorität, der über eine höhere Masse verfügt (z. B. Deuterium vor Hydrogenium). Sind Ordnungszahlen und Massen identisch, so hat der Substituent Priorität, der in direkter Verbindung mit einem weiteren Atom mit der höheren Ordnungszahl steht. Notfalls müssen noch die "dritten Atome" herangezogen werden. Doppel- und Dreifachbindungen zählen für das beteiligte Atom oder die Atomgruppe doppelt bzw. dreifach (Abb. 4).
Für stereochemische Unterschiede der Liganden gilt: cis vor trans , Z vor E und R vor S .
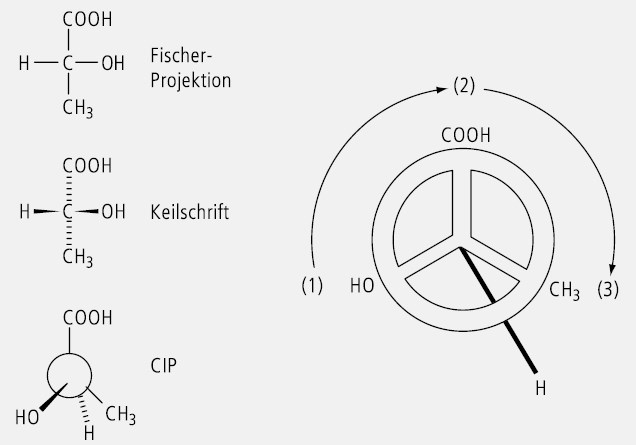
Um die absolute Konfiguration zu beschreiben, wird das Chiralitätszentrum so betrachtet, dass die Liganden 1, 2 und 3 in einer kreisförmigen Zählrichtung zum Beobachter zeigen und der Ligand 4 von ihm wegzeigt. Verläuft die Bewegung im Uhrzeigersinn, so liegt eine R -Konfiguration vor. Verläuft sie gegen den Uhrzeigersinn, so handelt es sich um eine S -Konfiguration.
Zum leichteren Verständnis kann man sich ein Lenkrad mit drei Speichen vorstellen, an deren Enden die Liganden 1 bis 3 sitzen, während die Lenksäule zum 4. Liganden führt (Abb. 5).
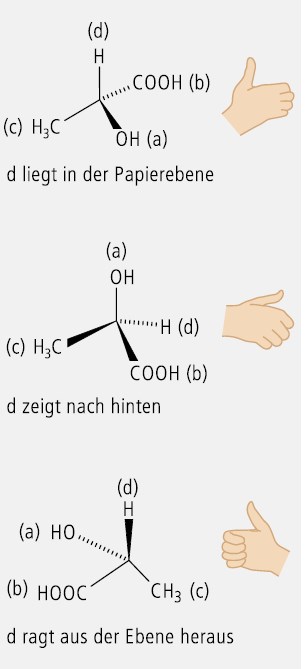
Am einfachsten lässt sich die R - oder S -Konfiguration mit der "Rechte-Hand-Regel " ermitteln. Sie basiert ebenfalls auf den CIP-Prioritätsregeln, benutzt als absolute Chiralitätsobjekte die Hände und ist auch bei komplizierten chiralen Strukturen anwendbar:
Zuordnung der CIP-Prioritäten a, b, c und d für die unterschiedlichen Substituenten an einem tetraedrischen Kohlenstoff- oder Hetero-Atom.
Der Daumen der rechten Hand deutet in Richtung d und stellt die C-d-Bindung dar.
Die Finger werden in Richtung der absteigenden Priorität der drei Substituentenpaare a-b, b-c und c-a gekrümmt, wobei der kürzeste Weg zu nehmen ist.
Gelingt diese Operation nicht, so ist die linke Hand zur Konfigurationsermittlung zu benutzen.
Gelingt es, die Finger der rechten Hand in Richtung der absteigenden Priorität zu krümmen, so liegt eine R-Konfiguration vor. Benötigt man dafür die linke Hand, so hat man eine S-Konfiguration ermittelt.
Abbildung 6 zeigt die Anwendung der Rechte-Hand-Regel am Beispiel der R-Milchsäure.
Das CIP-System ist auch bei zyklischen oder überbrückten Molekülen und bei komplexen Molekülen mit mehreren bis vielen Chiralitätszentren anwendbar.
sn-Nomenklatur
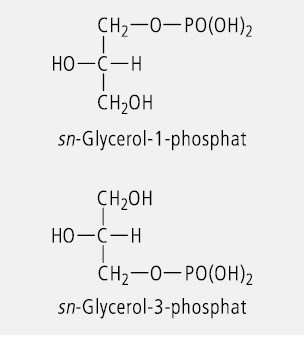
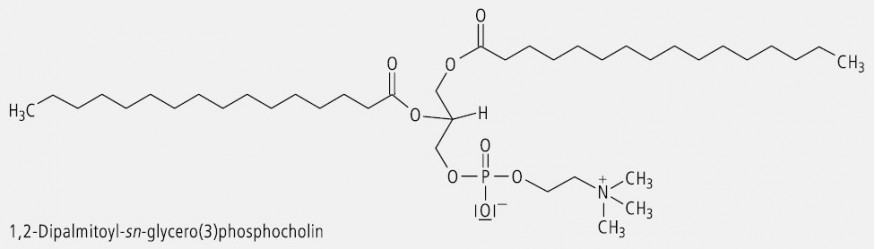
Glycerol (Glycerin) ist ein prochirales Molekül. Wird die mittelständige OH-Gruppe substituiert, so ändert sich daran nichts. Wird dagegen die OH-Gruppe in der relativen Position 1 oder der relativen Position 3 durch Substitution oder Austausch der funktionellen Gruppe verändert, dann entsteht ein chirales Molekül. Das ist z. B. bei den natürlichen Glycerophospholipiden der Fall. In der Lipidnomenklatur wird hierbei zur stereochemischen Charakterisierung das "stereospecific numbering system" (sn) verwendet. Dazu wird das Molekül so angeordnet, dass die freie oder substituierte Hydroxylgruppe in Position 2 nach links weist und der geminale Wasserstoff nach rechts. Das darüber stehende C-Atom wird dann als C(1) bezeichnet und das darunter stehende als C(3). Das Prinzip der sn-Nomenklatur ist in Abbildung 7 am Beispiel des Glycerolphosphats verdeutlicht.
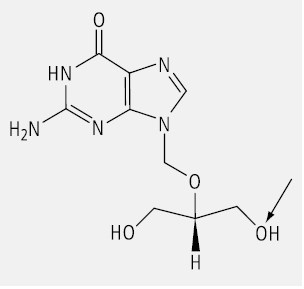
Ein prochirales Gycerol-Derivat stellt das Virustatikum Ganciclovir dar (Abb. 8). Als Beispiel für einen Arzneistoff, der nach der sn-Nomenklatur zu benennen ist, kann das Colfoscerolpalmitat (Abb. 9) dienen; es ist eine Komponente der Surfactans, die man zur Behandlung des Atemnotsyndroms von Neugeborenen (Infant Respiratory Distress Syndrom, IRDS) erfolgreich einsetzt.
Heteroatome als Chiralitätszentren
Außer den Verbindungen mit "asymmetrischem Kohlenstoff" können auch Moleküle mit anderen vierbindigen asymmetrisch substituierten Atomen wie Stickstoff, Schwefel und Phosphor zentrale Chiralität aufweisen. Das gleiche Phänomen bei Silicium, Zinn, Germanium und koordinierten Übergangsmetallen ist für den Bereich der Arzneistoffe derzeit nicht relevant.
Chirale Stickstoffverbindungen
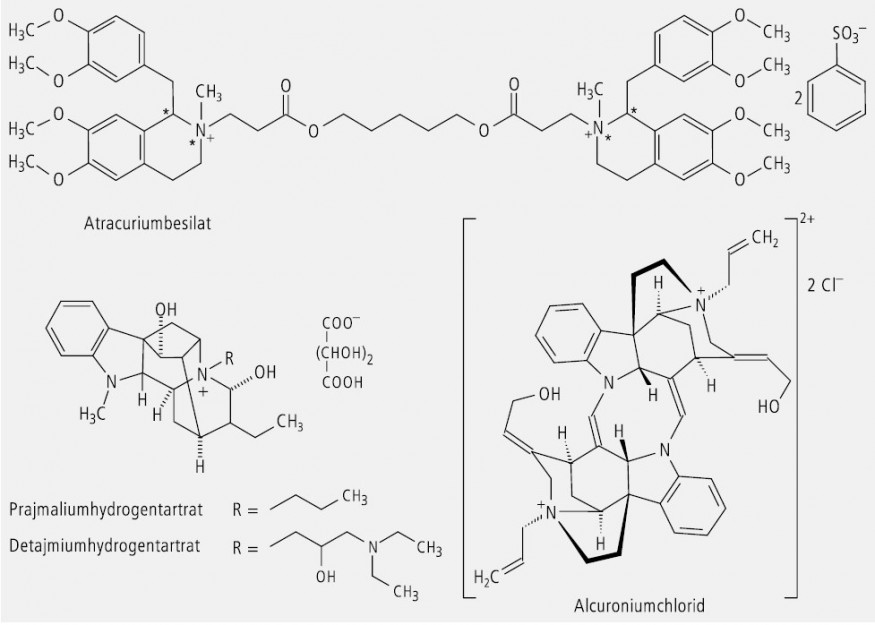
Quartäre Ammoniumverbindungen mit vier verschiedenen Liganden sind erwartungsgemäß chiral und lassen sich in Enantiomere auftrennen. Als Vertreter dieser Eigenschaft findet man u. a. die stabilisierenden Muskelrelaxanzien Atracurium-besilat und Alcuronium-chlorid und die Antiarrhythmika Prajmalium- und Detajmium-hydrogentartrat (Abb. 10). Die Letzteren sind durch die Besonderheit gekennzeichnet, dass der Stickstoff in Position 4 wegen seiner Fixierung in einem starren Ringsystem bereits ein Chiralitätszentrum darstellte, bevor er durch Alkylierung quartär wurde.
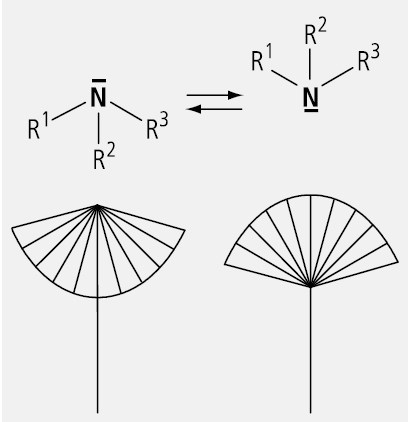
Tertiäre Amine mit drei verschiedenen Resten sind theoretisch ebenfalls zentrochirale Moleküle. Als vierter Ligand zählt das freie Elektronenpaar. Praktisch sind jedoch wegen der schnellen (pyramidalen) Inversion, die über einen planaren Übergangszustand verläuft, keine Enantiomere isolierbar. Man bezeichnet dieses Phänomen als Schirmeffekt (Abb. 11).
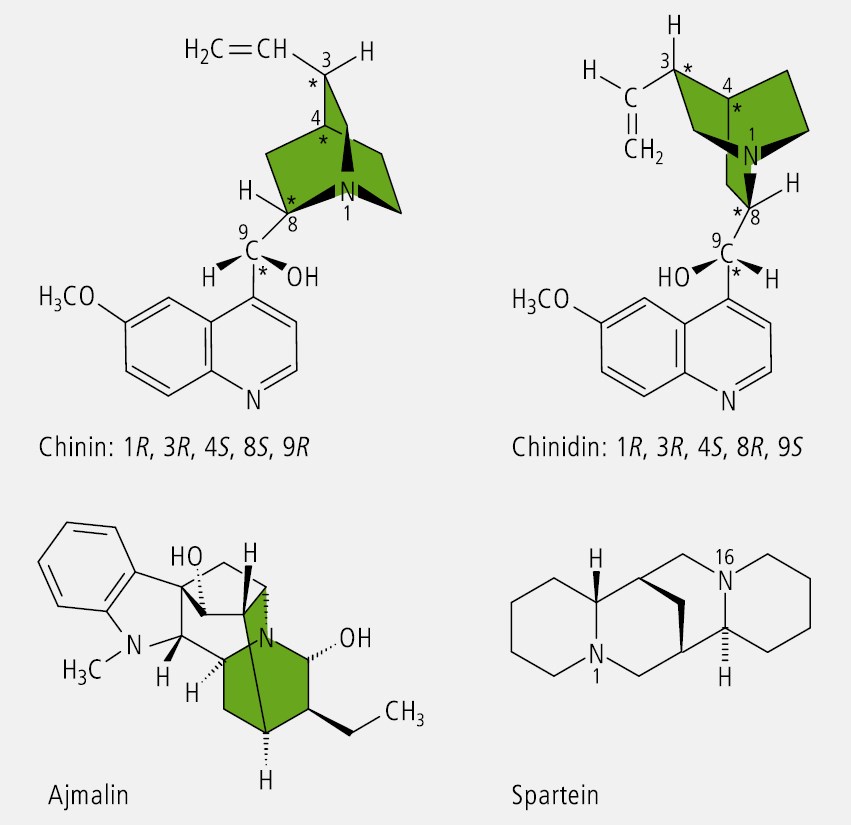
Bei Verbindungen, in welchen der tertiäre Stickstoff in ein starres oder überbrücktes Ringsystem eingebaut ist, kann keine Inversion erfolgen. Er stellt ein Chiralitätszentrum dar, wenn die drei an ihn gebundenen Reste verschieden sind, wie es auf die Chinaalkaloide (Chinin, Chinidin u. a.) und das Rauwolfia-Alkaloid Ajmalin zutrifft ( Abb. 12).
Auch das Antiarrhythmikum l‑Spartein (Abb. 12), das Hauptalkaloid des Besenginsters (Cytisus scoparius syn.Sarothamnus scoparius), das ein überbrücktes Ringsystem aus zwei Chinolizidinen darstellt, ist ein Beispiel für die Chiralität tertiärer Amine.
Chirale Phosphorverbindungen
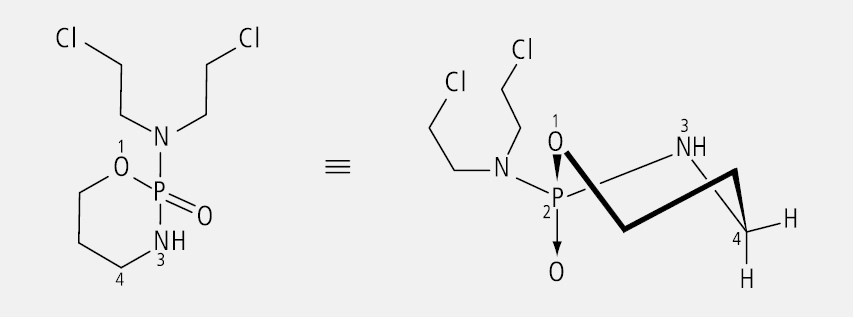
Das alkylierende Zytostatikum Cyclophosphamid ist wegen des asymmetrisch substituierten Phosphors eine chirale Verbindung (Abb. 13). Es wird immer noch als Racemat eingesetzt, obwohl gezeigt werden konnte, dass das (–)-Enantiomer eine deutlich höhere Zytotoxizität aufweist als das (+)-Enantiomer, was auf pharmakokinetischen Unterschieden beider Enantiomere beruht.
Chirale Schwefelverbindungen
Sofern sie zwei unterschiedliche Substituenten tragen, sind Sulfoxide, Sulfinsäureester, Sulfoniumsalze und Sulfine chiral. Eine Inversion der zusammen mit dem Sauerstoff dreifach substituierten Schwefelverbindungen ist beim Vergleich mit den dreifach unterschiedlich substituierten Stickstoffverbindungen (sprich tertiären Aminen) nicht zu beobachten. Sie erweisen sich als konfigurativ stabil.
Naturstoffe mit asymmetrisch substituiertem Schwefel sind Alliin und Allicin. Alliin ist ein genuiner Inhaltsstoff des Knoblauchs (Allium sativum). Durch das Enzym Alliin-Lyase wird es zum Allicin (Abb. 14) metabolisiert, das den essenziellen Faktor des bekannten Knoblauchgeruchs darstellt. Allicin enthält als Monoxid des Diallyldisulfids nebeneinander ein chiral substituiertes und ein achirales Schwefelatom.
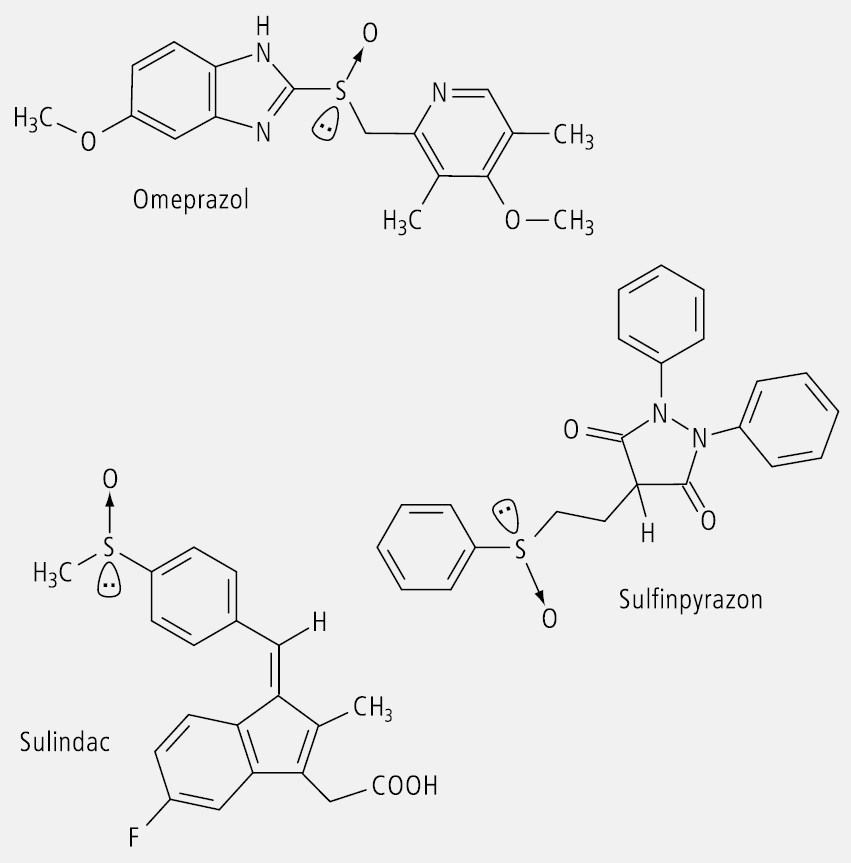
Zu den Arzneistoffen, die Chiralität am Schwefel als Zentrum aufweisen, gehören die Ulkus-Therapeutika Omeprazol und Verwandte, das nicht steroidale Antirheumatikum Sulindac und das Urikosurikum Sulfinpyrazon (Abb. 15).
Arzneistoffe mit zwei Chiralitätszentren
Wenn ein acyclisches Molekül mit unsymmetrischer Konstitution n verschiedene Chiralitätszentren enthält, sind von ihm 2n Stereoisomere zu erwarten. Moleküle mit symmetrischer Konstitution verfügen über weniger als 2n Stereoisomere.
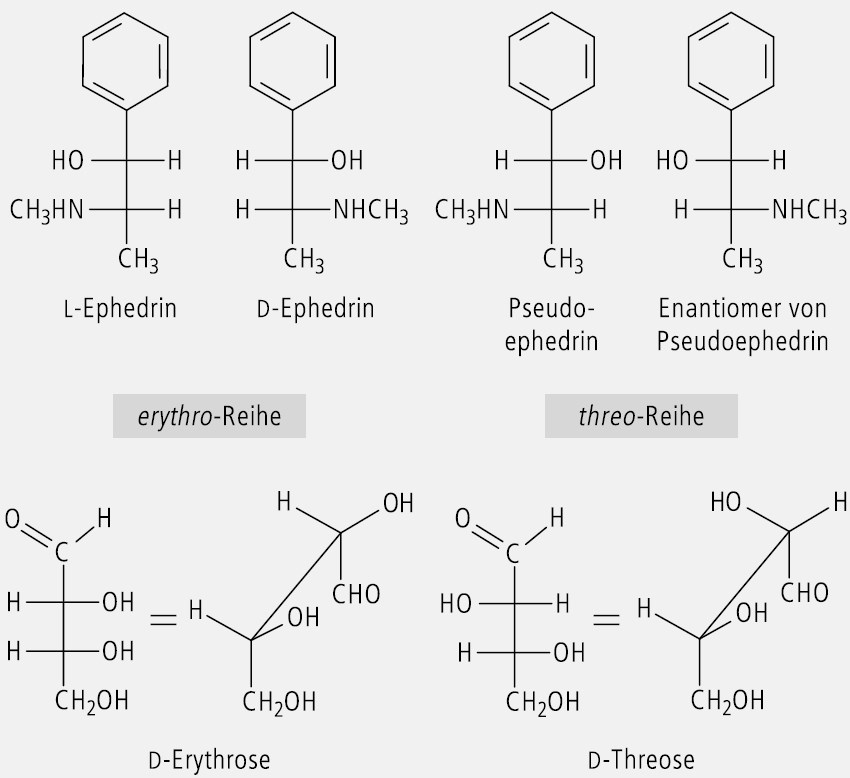
Offenkettige Verbindungen mit zwei benachbarten Chiralitätszentren, die zwei gleiche oder vergleichbare Substituenten (z. B. OH und OH oder OH und NH2) tragen, werden zur Kennzeichnung ihrer relativen Konfiguration mit der Threose und der Erythrose (Abb. 16) verglichen. Man spricht dann von einer threo-Form und einer erythro-Form.
In Abbildung 16 ist der Vergleich von Ephedrin und Pseudoephedrin mit Erythrose und Threose dargestellt. l-Ephedrin und d-Ephedrin sind Enantiomere. Ephedrin und Pseudoephedrin sind Diastereomere. Ein erythro-Isomer ist nicht enantiomer zu seinem threo-Isomer, sondern sie sind Diastereomere.
Moleküle mit symmetrischer Konstitution
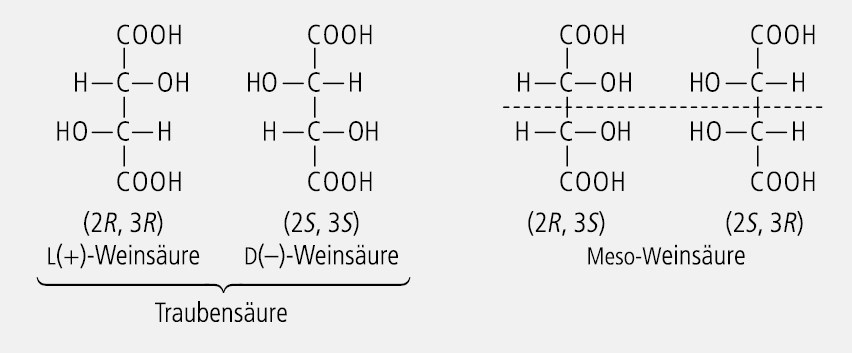
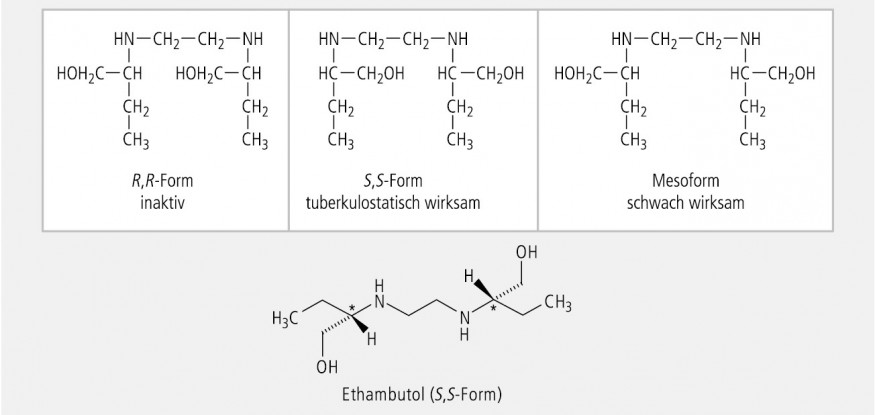
Von Verbindungen mit zwei konstitutionell spiegelbildlichen Asymmetriezentren, die zwei identisch verbundene, identisch substituierte Atome enthalten, existieren nicht 22 = 4, sondern nur drei Stereoisomere, nämlich die zwei Enantiomere (R,R und S,S), sowie die Mesoform (R,S), die aufgrund der molekularen Symmetrie mit der (S,R)-Form identisch ist. Das klassische Beispiel für solche Moleküle ist die Weinsäure (Abb. 17).
Ein symmetrischer Arzneistoff mit zwei identisch asymmetrisch substituierten C-Atomen ist das Chemotherapeutikum Ethambutol. Wie bei der Weinsäure können nur drei Stereoisomere existieren (Abb. 18). Die tuberkulostatische Aktivität ist weitgehend dem rechtsdrehenden S,S-Enantiomer zuzuschreiben.
Pseudoasymmetrie
Wenn in einem spiegelsymmetrischen Molekül ein zentrales C‑Atom vier verschiedene Liganden trägt, von welchen zwei konstitutionell identisch aber entgegengesetzt konfiguriert sind, so bezeichnet man es als pseudoasymmetrisch.
Durch die unterschiedliche Konfiguration der Chiralitätszentren in den beiden konstitutionell übereinstimmenden Liganden wird es zwar definitionsgemäß asymmetrisch, wegen der molekularen Symmetrie bleibt es aber ein achirales Zentrum.
Das Phänomen soll an einer linearen Verbindung und an einem überbrückten Heterocyclus erläutert werden.
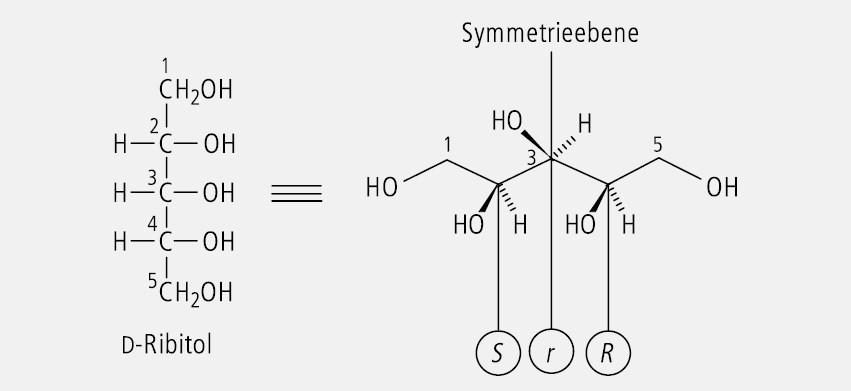
d-Ribitol, das in Abbildung 19 einmal in Form der Fischer-Projektion und einmal als stereochemische Zickzack-Formel dargestellt ist, besitzt auf den ersten Blick zwei Chiralitätszentren. C(2) ist S-konfiguriert, C(4) ist R-konfiguriert. Sie verhalten sich zueinander wie Bild und Spiegelbild. Damit besitzt das Molekül eine Spiegelebene, die durch C(3) verläuft. C(3) ist deshalb nicht asymmetrisch, sondern pseudoasymmetrisch substituiert. Seine Konfiguration wird mit (s) ausgedrückt. Die Entscheidung zwischen (r) und (s) ist mithilfe der CIP-Regeln zu treffen, wonach (R) vor (S) Priorität genießt.
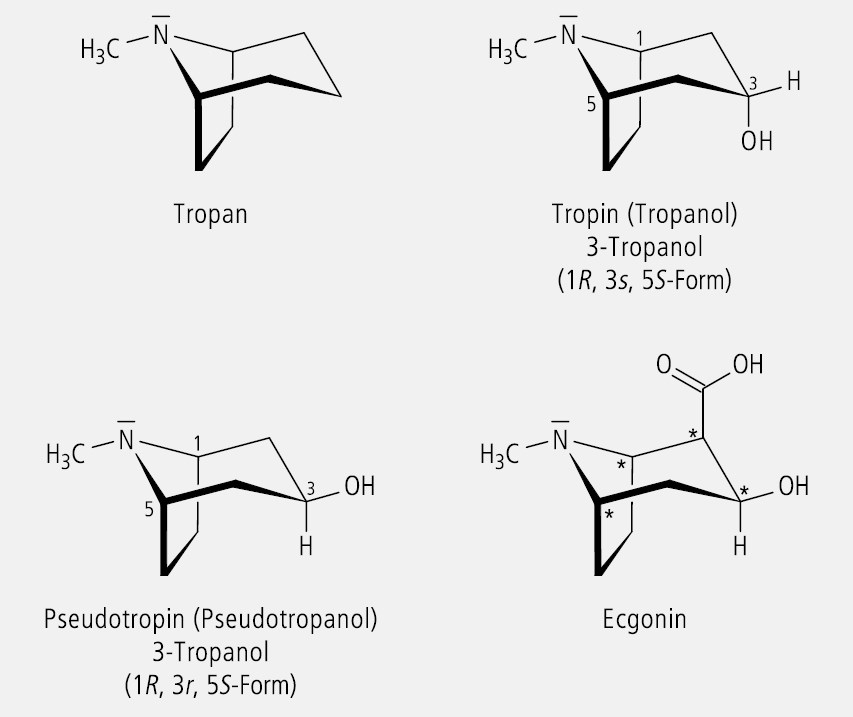
Tropan (Abb. 20), das Grundgerüst der Tropan-Alkaloide, ist ein überbrücktes Piperidin bzw. Pyrrolidin, in dem der Stickstoff sterisch fixiert ist. Die N-ständige Methylgruppe kann, bezogen auf den teilfixierten Piperidinring, entweder equatorial oder axial angeordnet sein. Damit existieren theoretisch zwei Stereoisomere, die sich aber wegen der rasch verlaufenden pyramidalen Inversion am Stickstoff nicht getrennt isolieren lassen.
Tropan enthält in den Positionen 1 und 5 zwei Chiralitätszentren entgegengesetzter Konfiguration. Wird die Position 3 hydroxyliert, so kann die OH-Gruppe eine axiale oder equatoriale Position einnehmen. Dadurch resultieren das Tropin (= Tropinol) und das Pseudotropin (= Pseudotropinol). Das C-Atom 3 ist infolge der molekularen Symmetrie pseudo-asymmetrisch (Abb. 20). Nach den CIP-Regeln (R > S) ergibt sich für Tropin eine 3(s)- und für Pseudotropin eine 3(r)-Konfiguration. Tropin ist der Alkanolamin-Teil des Ester-Alkaloids Hyoscyamin und dessen Racemat Atropin. Racemat bezieht sich auf den chiralen Säure-Teil des Esteralkaloids, die Tropasäure. Ecgonin, der Alkanolamin-Teil des Cocains ist ein in Position 2 substituiertes Pseudotropin (Abb. 20). Durch diese Substitution entsteht ein neues Chiralitätszentrum und es wird die Symmetrie des Pseudotropins aufgehoben. Nun liegt ein Molekül mit vier "echten" asymmetrisch substituierten C-Atomen in den Positionen 1, 2, 3 und 5 vor.
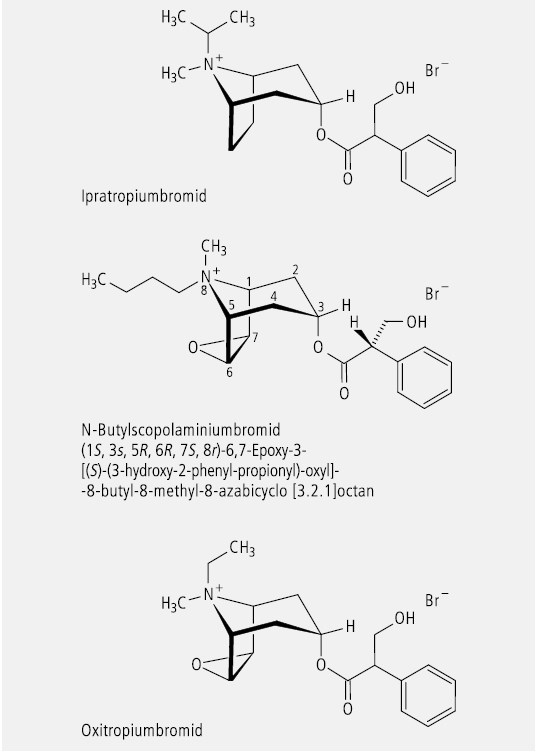
Wenn der Stickstoff im Tropin durch Alkylierung oder Oxidation quartär wird und sich die beiden Liganden strukturell unterscheiden, dann existieren zwei Stereoisomere mit unterschiedlichen physikalischen und spektroskopischen Eigenschaften. Molekulare Belege von jeweils einer Form sind die Arzneistoffe Ipratropiumbromid, N-Butylscopolaminiumbromid und Oxitropiumbromid (Abb. 21).
Axiale Chiralität und Atropisomerie
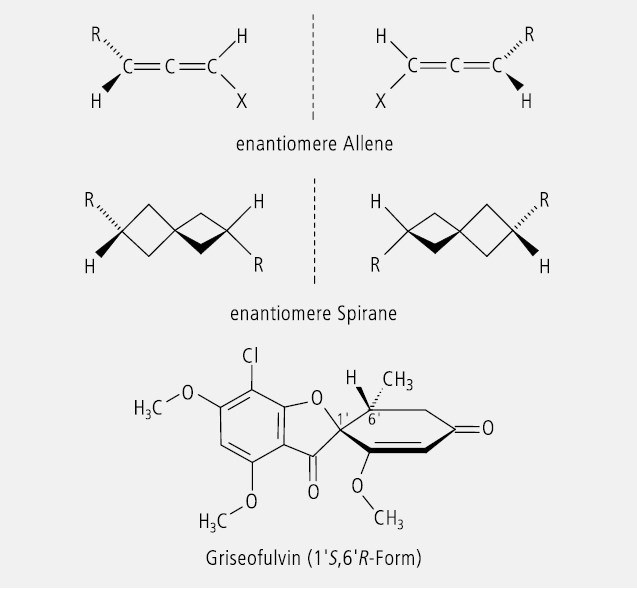
Verbindungen können auch dann chiral sein, wenn sie statt eines Asymmetriezentrums eine Chiralitätsachse enthalten. Das asymmetrisch substituierte C- oder Heteroatom ist nicht die einzige oder alleinige Ursache für Chiralität, obwohl sie unter den Arzneistoffen dominiert und sich nur wenige Beispiele für solche mit axialer Chiralität finden lasen. Chiral sind in bestimmter Weise substituierte Allene, Spirane und Biphenyl-Derivate. Die Substitution besteht in jeweils zwei ungleichen Liganden am Achsenende, bei Spiranen auch an anderen Positionen (Abb. 22).
Einer der seltenen Arzneistoffe mit chiraler Spiranstruktur ist das Griseofulvin (Abb. 22), mit einer beachtenswert hohen spezifischen Drehung: 354° – 364° in Dimethylformamid.
Die Substituenten H und R liegen paarweise in zueinander senkrechten Ebenen. Dadurch resultieren zwei spiegelbildliche Enantiomere, die nicht zur Deckung zu bringen sind. Verlängert man das Cumulen oder erweitert man das Spiran zu einem Bi-, Tri- Tetra-Spiran usw., dann tritt abwechselnd E/Z -Isomerie und Enantiomerie auf, weil die Substituenten abwechselnd in der gleichen und in zueinander senkrecht stehenden Ebenen liegen (Abb. 23).
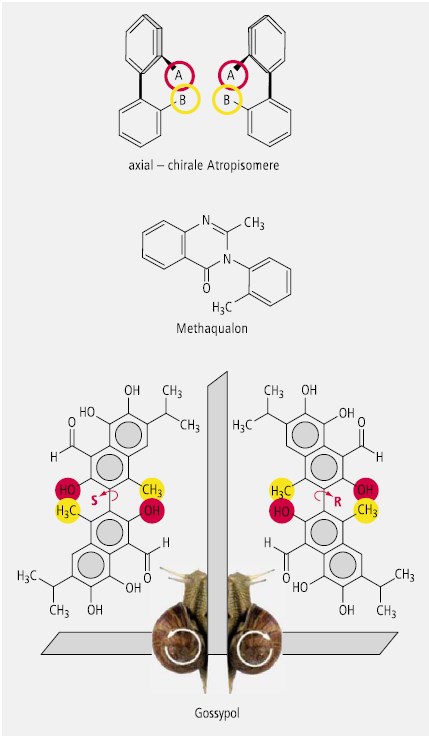
Biphenyl-Derivate mit axialer Chiralität werden nach einem Vorschlag von Richard Kuhn als Atropisomere bezeichnet. Biphenyle sind dann axial-chiral, wenn sie in zwei ortho-Stellungen sperrige Substituenten tragen, die eine Rotation um die Achse verhindern, welche die beiden Benzolringe miteinander verbindet (Abb. 24).
Ein atropisomerer Wirkstoff ist das aromatische Triterpen Gossypol (Abb. 24), das als gelbes Pigment in den Baumwollsamen (Stammpflanze Gossypium spp.) vorkommt, und als Fertilitätshemmer für Männer getestet wurde.
Auch Verbindungen mit gehinderter Rotation, bei denen zwei Aromaten über ein Heteroatom miteinander verbunden sind, können als Atropisomere betrachtet werden. Als Beispiel sei das Methaqualon genannt (Abb. 24).
Bei einigen NSAIDs wie Diclofenac und Meclofenaminsäure liegen theoretisch auch Atropisomere vor, die sich aber bei Raumtemperatur nicht in ihre Enantiomere trennen lassen. Gleiches gilt für Diflunisal, das ein echtes Biphenyl-Derivat darstellt.
Planare Chiralität
Planare Chiralität findet man beispielsweise bei substituierten para- und meso-Cyclophanen, beim trans-Cycloocten und bei Ansa-Verbindungen. Der Terminus ist allerdings problematisch, da die Chiralität dieser Verbindungen erst aus der räumlichen Partialstruktur entsteht, die sich über oder unter dem planaren Molekülteil ausbreitet.
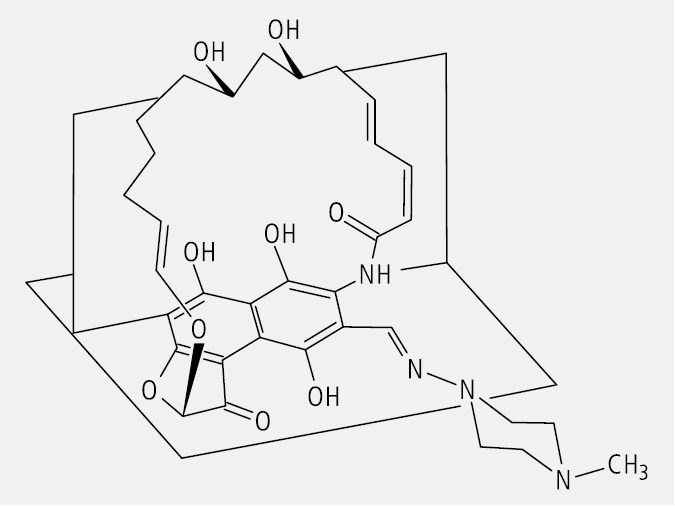
Arzneistoffe mit planarer Chiralität sind das Rifampicin (Abb. 25) und seine Verwandten.
Helicität
Helikale, d. h. schraubenförmige Strukturen, die wir von makroskopischen Naturformen wie Schnecken oder gewundenen Pflanzen kennen, treten auch im molekularen Bereich auf, so bei Oligo- und Polypeptiden, Polysacchariden, Nucleinsäuren (DNA und RNA) und bestimmten Metallkomplexen. Von plus und minus abgeleitet, wird die rechtsgängige Schraubenform als p-Helix bezeichnet und die Linksschraube als m-Helix.
Peptid-Antibiotika, die eine helikale Struktur in Bakterienzellmembranen ausbilden, sind die Gramicidine. Glucagon, der funktionelle Antagonist des Insulins, bildet in wässriger Lösung eine α-Helix aus.
Diastereomere
Diastereomeren-Moleküle können achiral oder chiral sein. Wenn zwei (oder mehrere) Stereoisomere nicht wie Bild und Spiegelbild erscheinen, verhalten sie sich wie Diastereomere. Sie lassen sich nicht, wie es bei den Enantiomeren der Fall ist, durch eine Spiegeloperation ineinander überführen.
Chirale Diastereomere bestehen aus Enantiomerenpaaren. Diastereomere haben unterschiedliche physikalische, chemische und spektroskopische Eigenschaften.
Achirale Diastereomere findet man bei Verbindungen, die eine Doppelbindung enthalten (außer C=C- auch C=N- und N=N-). Ideale Muster sind die Maleinsäure und die Fumarsäure, sowie die Ölsäure und die Elaidinsäure (Abb. 26). Achirale Diastereomere unterscheiden sich ebenfalls stark in ihren physikalischen, chemisch-reaktiven und physiologischen Eigenschaften. Die Z-konfigurierte Maleinsäure mit einem Schmelzpunkt von 132 °C zeigt eine geringere Stabilität und eine höhere Reaktivität als die E-konfigurierte Fumarsäure mit einem Schmelzpunkt von 286 °C. Die bei Raumtemperatur flüssige Ölsäure ist als Nahrungsbestandteil physiologisch wertvoll. Die Elaidinsäure weist einen Schmelzpunkt von 51 °C auf und gehört zu den trans -Fettsäuren, die bei der partiellen Hydrierung natürlicher Fettsäuren entstehen und physiologisch bedenklich sind.
Die Deskriptoren cis und trans sind hier nicht immer eindeutig. Wenn etwa vier verschiedene Substituenten an der Doppelbindung haften, wird die Zuordnung schwierig oder unmöglich. Deshalb verwendet man heute die Deskriptoren Z und E (die Anfangsbuchstaben von "zusammen" und "entgegen"), die auf den CIP-Regeln beruhen.
Bei ringförmigen Verbindungen mit ungerader Anzahl an Ringatomen stellt das Z-Isomer eine Mesoform mit R,S-Konfiguration dar und ist deshalb achiral. Das E-Isomer existiert in einer R,R-Form und einer S,S-Form, die ein Enantiomerenpaar bilden. Bei disubstituierten Ringen mit gerader Anzahl an Ringatomen, wie es beim Cyclohexan der Fall ist können bei zwei unterschiedlichen Substituenten verschiedene Isomere auftreten. Bei 1,1-Disubstitution existiert keine Chiralität, da sich die beiden anderen Substituenten am gleichen C-Atom infolge der molekularen Symmetrie wie zwei gleiche Reste verhalten. Bei 1,4-Disubstitution ist cis/trans-Isomerie zu beobachten, aber keine Chiralität wegen der Symmetrieebene. Bei 1,2- und 1,4-Disubstitution tritt je nach Substitutionsmuster cis/trans -Isomerie und Chiralität auf. Man erhält vier Stereoisomere, jeweils ein trans- und ein cis-Enantiomerenpaar.
Topizität
Dieser Begriff ist nicht nur für einen speziell interessierten Chemiker von Bedeutung, sondern bildet auch Grundlage für das Verständnis der Biotransformation von Arzneistoffen. Die Topizität befasst sich mit der räumlichen (topischen, stereochemischen) Beziehung von Substituenten in einer Grundstruktur. In Abhängigkeit von der Umgebung der Substituenten spricht man von homotop, enantiotop und diastereotop. Die zugehörigen Verläufe werden Homotopizität oder Homotopie, Enantiotopizität oder Enantiotopie und Diastereotopizität oder Diastereotopie genannt.
Homotopie
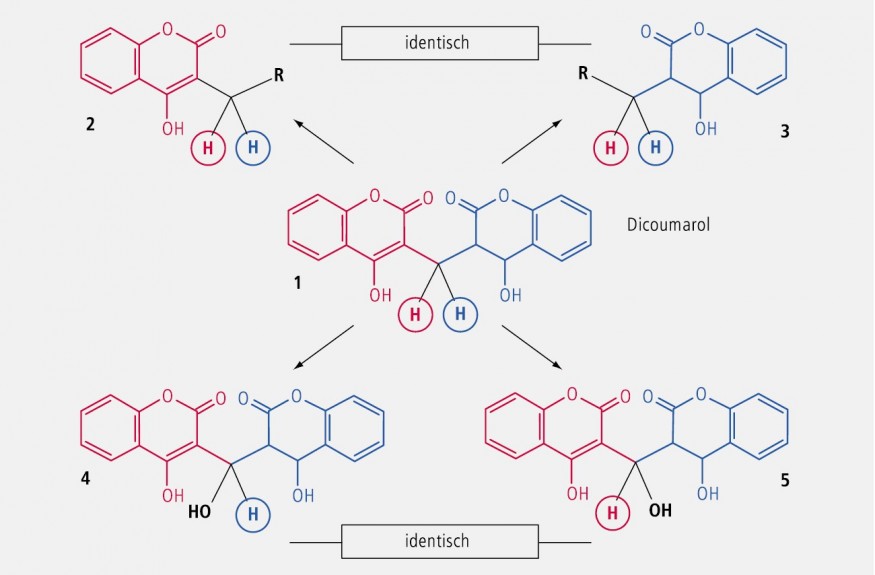
Zwei identische Substituenten an einem tetraedrischen C-Atom, die von zwei anderen, aber ebenfalls identischen Substituenten am selben C-Atom umgeben sind, bezeichnet man als homotop. Sie sind stereochemisch äquivalent und daher nicht unterscheidbar. Charakteristisch für homotope Gruppen ist der Erhalt der Achiralität beim Ersetzen einer der beiden Gruppen.
In Abbildung 27 ist der Vorgang am Beispiel Dicoumarol erläutert: Wird der blau gezeichnete Molekülteil durch einen Substituenten R ersetzt, so resultiert die Verbindung 2 • Ersetzt man den rot gezeichneten Molekülteil durch den gleichen Rest R, so entsteht die Verbindung 3 • Wegen der fehlenden Chiralität sind beide Produkte (2 und 3) identisch. Analoges geschieht beim Ersatz eines H-Atoms der Methingruppe des Dicoumarols, d. h. die Metaboliten (4 und 5) sind ebenfalls identisch.
Enantiotopie und Prochiralität
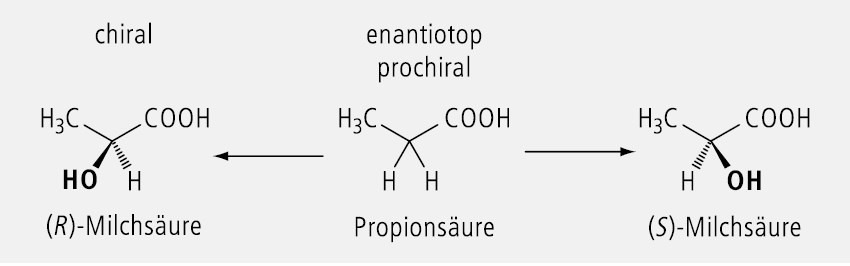
Zwei identische Substituenten an einem tetraedrischen C-Atom, die von zwei unterschiedlichen Substituenten am selben C-Atom flankiert sind, bezeichnet man als enantiotop. Wird einer der beiden identischen Substituenten durch eine beliebige achirale Gruppe ausgetauscht, so entsteht ein asymmetrisch substituiertes C-Atom, d. h. es wird ein Chiralitätszentrum gebildet. Ein C-Atom mit zwei enantiotopen Gruppen nennt man deshalb prochiral. Der wechselweise Austausch führt zu einem Enantiomeren-Paar, d. h. es entsteht ein Racemat.
Ein praktisches Beispiel ist in Abbildung 28 zu finden: Aus der enantiotopen Propionsäure entsteht das Racemat der Milchsäure.
Im Gegensatz zu homotopen Gruppen sind enantiotope Gruppen stereochemisch unterscheidbar. Nach der CIP-Nomenklatur werden sie als pro-R und pro-S bezeichnet. Die beiden Substituenten am prochiralen C-Atom unterscheiden sich durch ihre topografische Position im Molekül. Stereospezifische Enzyme und Rezeptoren sind in der Lage, beide Gruppen zu unterscheiden. Dadurch wird verständlich, dass bei der Biotransformation prochiraler Arzneistoffe ausschließlich oder bevorzugt ein Enantiomer als Metabolit entsteht und nicht ein Racemat.
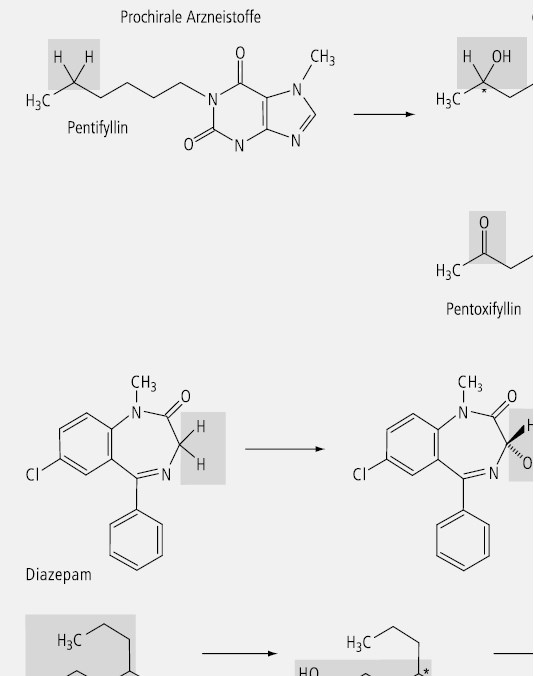
Beispiele prochiraler Arzneistoffe, die enantioselektiv metabolisiert werden, sind Pentifyllin, Diazepam und Valproinsäure (Abb. 29). Die enantioselektive Hydroxylierung des prochiralen Pentifyllins führt zu einem chiralen Carbinol, das dann in einem weiteren Oxidationsschritt zum wiederum prochiralen Pentoxifyllin metabolisiert wird. Besonders interessant verläuft die enantioselektive Metabolisierung bei Phenylbutazon und bei Phenytoin.
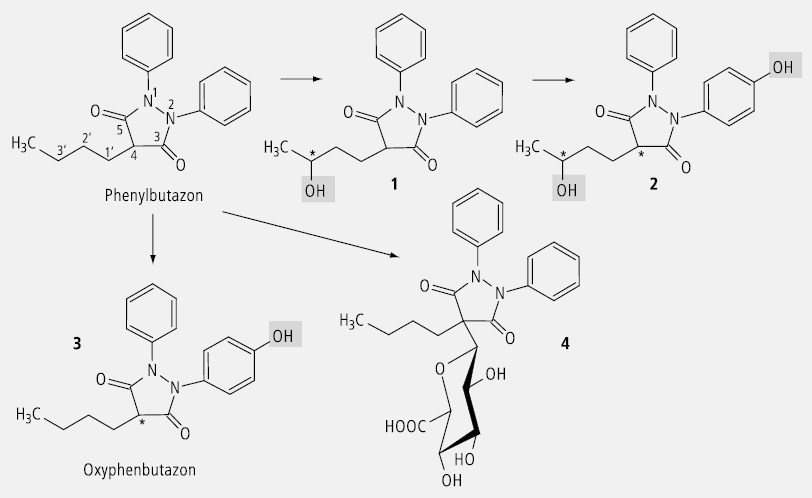
Phenylbutazon (Abb. 30) wird durch die Hydroxylierung in der Seitenkette chiral (1). Durch die Hydroxylierung eines Benzolringes erhält auch ein C-Atom (C4) im Butazolring Asymmetrie (2). Es entstehen Diastereomere. Die alleinige Hydroxylierung eines Benzolringes führt zum Enantiomer 3 , und die Glucuronidierung am C4 lässt wegen der Chiralität der Glucuronsäure ein Diastereomer entstehen (4).
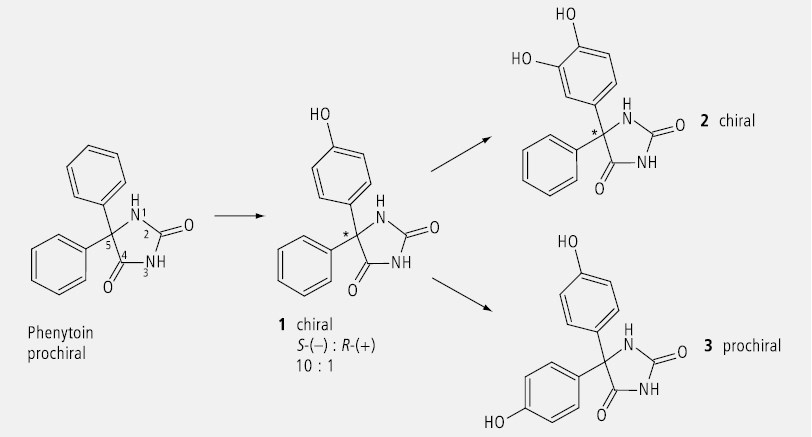
Auch Phenytoin (Abb. 31) wird als prochirale Verbindung durch Hydroxylierung an einem Benzolring chiral (1), wobei das Verhältnis vom S-Enantiomer zum R-Enantiomer 10 zu 1 beträgt. Eine weitere Hydroxylierung am selben Aromaten ändert daran nichts (2). Wird dagegen jeder der beiden Benzolringe in äquivalenter Position hydroxyliert, so resultiert wieder ein prochirales Molekül (3).
Dass die Betrachtung des prochiralen Phenytoin unterschiedliche pro-R- und pro-S-Seiten ergibt, soll durch die skizzenhafte Abbildung 32 verdeutlicht werden. Von oben betrachtet ist die Atomfolge von links nach rechts – C– NH– CO– , von unten in gleicher Weise betrachtet ergibt sich die Atomfolge – C– CO– NH–.
Diastereotopizität
Ist bei dem voranstehenden Ablauf die neu eintretende Gruppe selbst chiral, so werden Diastereomeren gebildet.
Zwei identische Substituenten an einem tetraedrischen C-Atom, die von zwei unterschiedlichen Substituenten am gleichen C-Atom flankiert sind, wobei der eine ein Chiralitätszentrum enthält, bezeichnet man als diastereotop. Im Unterschied zu homotopen und enantiotopen Verbindungen können diastereotope Verbindungen durch keine Symmetrieoperation zur Deckung gebracht werden.
Stereochemische Betrachtung kondensierter Ringsysteme
Bei bi- bis polycylischen Ringsystemen kann auch eine cis/trans -Isomerie auftreten. Die Bezeichnung der Verknüpfungsart richtet sich dabei nach der Stellung der Substituenten an den axialen Atomen bzw. an der Bindung, die den beiden Ringen gemeinsam angehört. Musterbeispiele sind die Steroide, welche die Grundgerüste der Sexualhormone, der Herzglykoside und der Gallensäuren darstellen.
In Abbildung 33 sind exemplarisch die Verknüpfungsarten von α-Gonan und β-Gonan aufgezeigt. Die strukturelle trans-trans-trans-Verknüpfung des α‑Gonans findet man bei den steroidalen Sexualhormonen (α‑Estran-, α-Androstan-, α‑Pregnan), die cis-trans-trans-Verknüpfung des β-Gonans bei den Gallensäuren (Cholan) und bei den Herzglykosiden.
Mit α und β wird die Lage des Wasserstoffatoms in Position 5 gekennzeichnet.
Gewinnung enantiomerenreiner Arzneistoffe
Die Gewinnung kann durch die Enantiomerentrennung eines Racemats oder durch stereoselektive Synthesen erfolgen. Man unterscheidet folgende Trennverfahren:
mechanische Enantiomerentrennung durch Handauslese,
Racematspaltung durch Überführung in Diastereomere,
kovalente Derivatisierung mit chiralen Reagenzien,
Bildung diastereomerer Komplexe bei der chromatografischen Enantiomerentrennung,
biochemische Racemattrennung,
- kinetische Trennung.
Synthese enantiomerenreiner Arzneistoffe
Prinzipiell existieren zwei Möglichkeiten: die Verwendung chiraler Bausteine aus der Natur und die asymmetrische (achirale) Synthese.
Die Natur stellt ein reichliches Angebot an enantiomeren Ausgangsstoffen zur Verfügung ("chiral pool"). Zu den wichtigsten Stoffklassen, die preiswert zu erhalten sind, gehören Aminosäuren, Zucker, Alkaloide und Terpene.
Eine achirale Synthese ist eine Reaktion, in der ein prochirales Molekül in ein chirales Molekül umgewandelt wird, wobei die stereoisomeren Produkte, die Enantiomere oder Diastereomere sein können, in ungleichen Mengen entstehen. Man unterscheidet:
interne chirale Induktion (Chiralitätsübertragung),
externe chirale Induktion, die in der asymmetrischen Synthese unter Verwendung chiraler Katalysatoren besteht, und
asymmetrische Synthesen mit Enzymen.
Dynamische Stereochemie
Moleküle mit feststehender Konstitution und Konfiguration können in dynamischer Weise verschiedene Konformationen (Konstellationen) einnehmen.
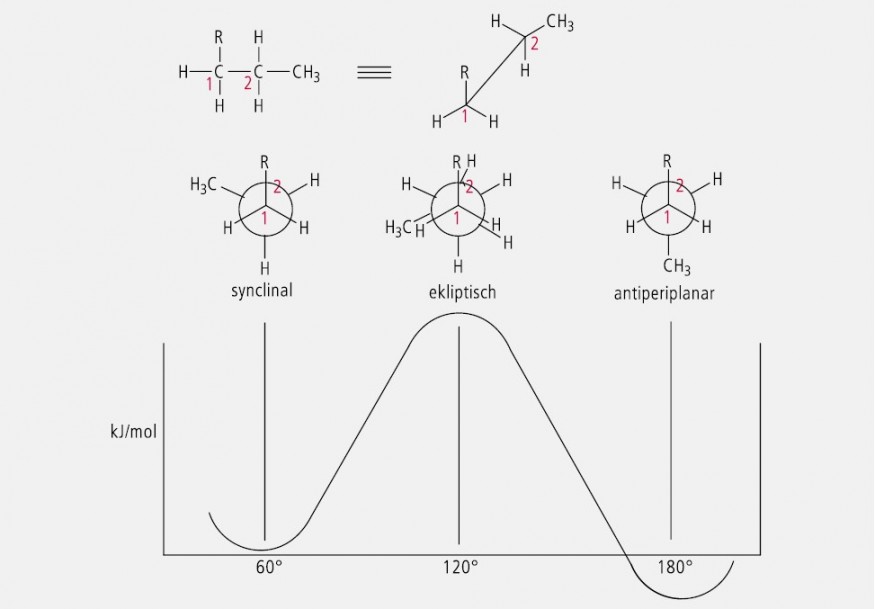
Als Konformationen werden die räumlichen Anordnungen von Atomen oder Gruppen eines Moleküls definierter Konfiguration bezeichnet, die durch Drehung um Einfachbindungen erzeugt und nicht deckungsgleich sind. Theoretisch sind bei einem Molekül gegebener Konfiguration sehr viele Konformationsisomere möglich, die man auch als Rotationsisomere oder Rotamere bezeichnet. Die verschiedenen Konformationsisomere unterscheiden sich durch verschiedene Energiegehalte mit Energiemaxima und Energieminima. Der Begriff Konformere wird üblicherweise für die real existierenden Konformationen verwendet, also solche mit einem Energieminimum. Drei exponierte Konformationen sind die synclinale, die ekliptische und die antiperiplanare Konformation, die am Beispiel der Verbindung R– CH2 – CH2 – CH3 mit den zugehörigen Energieniveaus in Abbildung 34 dargestellt sind.
Die drei verschiedenen Konformationen des Cyclohexans sind die Sesselform, die Wannenform und die Twistform (Abb. 35). Die energieärmste und damit stabilste Konformation ist die Sesselform. In ihr liegen sechs H-Atome axial (a), d. h. parallel zur dreizähligen Symmetrieachse, abwechselnd nach oben und nach unten gerichtet vor. Sechs H-Atome liegen equatorial (e) vor, d. h. sie sind zur mittleren Ebene des Ringes geneigt und somit näher an dieser Ebene als die axialen H-Atome. Die Sesselform kann durch Konformationsänderung in die Wannenform umklappen, wobei die axialen H-Atome zu equatorialen und die equatorialen H-Atome zu axialen werden. Große Substituenten am Cyclohexanring ordnen sich bevorzugt in equatorialer Lage an, wodurch eine höhere Stabilität erreicht wird.
Reaktionen am Chiralitätszentrum
Für den Ablauf einer Reaktion an einem Chiralitätszentrum bestehen vier Möglichkeiten:
1. Retention: Konfigurationserhalt durch eine SN i-Reaktion,
2. Inversion: Konfigurationsumkehr durch eine SN 2-Reaktion,
3. Racemisierung: Umwandlung einer enantiomerenreinen Verbindung in beide Enantiomere durch eine SN 1-Reaktion,
4. Verlust der Chiralität durch Eliminierungsreaktionen.
Als Retention wird eine Substitutionsreaktion bezeichnet, bei der die Konfiguration an einem Asymmetriezentrum erhalten bleibt. Ebenso kann man diesen Vorgang als stereokonservative Reaktion bezeichnen. Zu einer Retention gelangt man auch durch doppelte Inversion (vergleichbar mit der doppelten Verneinung im Sprachgebrauch).
Die Inversion ist eine Substitutionsreaktion, bei der es zur Umkehr der Konfiguration an einem Asymmetriezentrum kommt, sodass aus einer R -Form eine S-Form entsteht und umgekehrt. Das Paradebeispiel ist die Walden-Umkehr. Unter den chiralen Arzneistoffen ist die enzymatisch katalysierte Inversion der schwach wirkenden R-Form des Ibuprofen in die stark wirkende S-Form deshalb bemerkenswert, weil die Inversion in umgekehrte Richtung nicht erfolgt.
Eine Racemisierung kann bereits bei der Isolierung eines Naturstoffs eintreten. Pharmazeuten und Naturstoffchemiker denken dabei spontan an die Umwandlung des Hyoscyamins in das racemische Atropin. Bekannt ist auch der rasche Übergang des nicht teratogen wirkenden R-Thalidomids in das teratogene S-Thalidomid. Die Contergan®-Katastrophe vor 50 Jahren wäre deshalb nicht verhindert worden, wenn das Schlafmittel Contergan® R-Thalidomid statt des Racemates enthalten hätte. Übrigens sind beide Thalidomid-Enantiomere in ihrer hypnotischen Wirkung gleich stark.
Der Verlust der Chiralität ist häufig durch eine β-Eliminierung oder durch Oxidation einer chiralen Carbinolgruppe zu einer Carbonylgruppe bedingt. β-Eliminierungen sind durch die Abspaltung von Atomen oder Atomgruppen an zwei benachbarten C-Atomen gekennzeichnet, die zur Bildung einer Doppelbindung führt. Sie können als Umkehrung der Addition an eine C=C-Doppelbindung betrachtet werden. Regioselektivität und Stereochemie hängen dabei von der Art der Substituenten und Reagenzien sowie von den Reaktionsbedingungen ab.
Isomerisierungen
Unter einer Isomerisierung versteht man ganz allgemein die Überführung einer Verbindung in ein Isomer. Stereochemische Isomerisierung kann stattfinden an einem Chiralitätszentrum eines Enantiomers oder Diastereomers und an E/Z- bzw. cis/trans-Isomeren.
Bei Zuckern, zuerst bei der Glucose beobachtet, kann man eine kontinuierliche Änderung des optischen Drehwertes der Lösungen feststellen, bis ein konstanter Wert erreicht ist. Dieses Phänomen wird Mutarotation genannt (von lat. mutare = vertauschen) und beruht auf der Epimerisierung an einem Asymmetriezentrum. Darunter versteht man eine bestimmte Form der Isomerisierung, nämlich die Konfigurationsänderung an nur einem von mehreren asymmetrischen C-Atomen einer organischen Verbindung.
Bei E/Z- bzw. cis-trans-Umwandlungen ist eine hohe Energiebarriere zu überwinden, die jedoch stark vom Substitutionsmuster abhängt. Die Isomerisierung kann photochemisch, thermisch oder katalytisch erfolgen. E-Diethylstilbestrol, ein Estrogen-Agonist, wird unter physiologischen Bedingungen teilweise rasch in das Z-Isomer umgewandelt. Kompliziertere Vorgänge der E/Z-Umwandlung laufen bei der photochemischen und thermischen Umwandlung von Ergosterol in Vitamin D2 ab. Eine bekannte cis-trans-Umlagerung findet auch im Rhodopsin in der Netzhaut statt (wichtig für das Farbensehen).
Substitutionsreaktionen, die unter geeigneten Bedingungen zu chiralen Verbindungen führen, sind die nucleophile und die elektrophile Substitution am gesättigten C-Atom. Voraussetzung für die eine ist eine nucleophile Abgangsgruppe, für die andere eine elektrophile Abgangsgruppe. Radikalische Substitution am gesättigten C-Atom führt in der Regel zu Racematen.
Additionsreaktionen an C=C-Doppelbindungen
Bei der Addition an eine energiereiche C=C-Doppelbindung entstehen aus der π-Bindung zwei σ-Bindungen. Der stereochemische Verlauf hängt dabei ab vom Reagens, von den schon vorhandenen Substituenten und vom Lösemittel. Diese Reaktion kann man als Umkehrung der β-Eliminierung betrachten (s. o.).
Thermodynamik und Stereochemie
Ein Fall von dynamischer Stereochemie ist bei den axial-chiralen Atropisomeren zu beobachten. Bei allen Atropisomeren verschwindet die oft sehr große optische Aktivität, wenn durch Temperaturerhöhung die innere Beweglichkeit im Molekül zunimmt und die Energiebarriere, die bei niedriger Temperatur eine Rotation verhindert, überwunden werden kann.
Thermodynamisch zu betrachten sind auch die Wechselwirkungen mit Poteinen. Natürliche Proteine sind fast ausschließlich aus l-Aminosäuren aufgebaut und sind dadurch chirale Makromoleküle. Da sich Enantiomere in chiraler Umgebung wie zwei verschiedene Verbindungen verhalten, ist es leicht zu verstehen, dass enantiomere Arzneistoffe auch eine unterschiedlich starke Proteinbindung aufweisen und von Enzymen differenziert angegriffen werden (das gilt auch für andere Wirkstoffe wie Pheromone oder Riechstoffe). Erinnert sei in diesem Zusammenhang an die Möglichkeit der enzymatischen Racematspaltung, wobei eines der beiden Enantiomeren abgebaut ("aufgefressen") wird und das andere sich anreichert.
Es gibt zahlreiche Beispiele von chiralen Arzneistoffen, bei welchen
das eine Enantiomer stärker wirkt als das andere oder
beide Enantiomere in unterschiedlicher Weise wirken oder
das eine Enantiomer sogar entgegengesetzt wirkt wie das andere.
Unterschiedliche Wirkqualitäten können dazu führen, dass das eine Enantiomer toxisch ist, das andere aber nicht (z. B. Thalidomid, s. o.).
Nach Ariens werden die Enantiomere mit positiven Eigenschaften (gut, stark, weniger toxisch) als Eutomere und die mit negativen Eigenschaften (schlecht, schwach, toxisch) als Distomere bezeichnet. Den Quotienten von beiden nennt man Eudismisches Verhältnis.
Bei vielen Terpenen, die in ätherischen Ölen vorkommen, unterscheiden sich die Enantiomere olfaktorisch. So riecht z. B. das S-Carvon nach Kümmel und das R-Carvon nach Pfefferminze.
Enantiomere Pheromone unterscheiden sich ebenfalls in ihrer Wirkung. So lockt zum Beispiel die S-Form des Oleans, das von der Olivenfliege (Dacus oleae) produziert wird, das Männchen an, während die R-Form das Männchen vertreibt.
Nachwort
Das breit gefächerte Pharmaziestudium stellt hohe Anforderungen an die Studierenden. Konnten früher die Dozenten der einzelnen Fächer auch in der Lehre ihre Schwerpunkte setzen, wurde diese Freiheit durch die gültige Approbationsordnung eingeengt. Damit sollten eigentlich die Studierenden entlastet werden. Wenn man allerdings die vom Institut für medizinische und pharmazeutische Prüfungsfragen (IMPP) erarbeiteten Gegenstandskataloge anschaut, könnte man fast glauben, es sollen Spezialisten herangezüchtet werden. Da ich mir in meinem Fachgebiet der Medizinisch-Pharmazeutischen-Chemie ein Urteil erlauben kann und darf, muss ich dringend vor Übertreibungen warnen. Hinzu kommt, dass unter den MC-Fragen, die den Studierenden seit 1979 vorgelegt wurden, reichlich spitzfindige Formulierungen grassieren. Ich selbst könnte nicht alle Fragen eindeutig beantworten. Doch sollten die Prüflinge auf ihre Grundkenntnisse getestet und nicht durch hinterlistige Fragen abgestraft werden!
Ich habe mich bemüht, in diesem Aufsatz die für eine(n) Arzneimittelfachfrau(mann) relevanten Aspekte – wenn immer es geht, auf Arzneistoffe bezogen – darzustellen. Aus Platzgründen musste ich mich auf Wichtiges beschränken. Auf das Wesentliche beschränken sollten sich auch die Kolleginnen und Kollegen, die bei der Erstellung oder Änderung der Approbationsordnung, der Verfassung von Gegenstandskatalogen und vor allem bei der Auswahl und verbalen Gestaltung von MC-Fragen zuständig sind.
Autor
Prof. Dr. rer. nat. Dr. h. c. Hermann J. Roth, Friedrich-NaumannStr. 33, 76187 Karlsruhe
www.h-roth-kunst.com



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.