















- DAZ.online
- DAZ / AZ
- DAZ 46/2012
- Vom Wirkstoff zum ...
UniDAZ
Vom Wirkstoff zum Arzneimittel
EditorialLiebe Leserinnen, liebe Leser! Nach der Zulassung darf ein Arzneimittel, das während der klinischen Prüfung bei einer begrenzten Anzahl von Patienten eingesetzt wurde, in größerem Stil angewandt werden. Dabei können unerwünschte Arzneimittelwirkungen (UAW) auftreten, die in der klinischen Prüfung übersehen wurden, z. B. weil sie auf Patienten mit bestimmten Eigenschaften (Risikogruppen) beschränkt sind. So nahm der Hersteller das ordnungsgemäß zugelassene Antirheumatikum Rofecoxib (Vioxx®) im Jahr 2004 wieder vom Markt, weil es das Risiko für einen Herzinfarkt erhöhte. Ärzte und Apotheker sind verpflichtet, unerwünschte Arzneimittelwirkungen, die ihnen bekannt werden, ihren Arzneimittelkommissionen zu melden. Auch der Hersteller muss ein waches Auge auf seine Produkte haben und gegebenenfalls mit dem Rote-Hand-Brief über Arzneimittelrisiken informieren. Wir wünschen Ihnen eine interessante Lektüre und möchten Sie auch an die beiden ersten Teile dieser UniDAZ-Serie erinnern: 1. Teil: Von der Präklinik zur Phase-I-Studie. DAZ Nr. 25, S. 70– 76. 2. Teil: Klinische Prüfungen. DAZ Nr. 29, S. 61– 67. Ihre UniDAZ-Redaktion |
Die klinische Erprobung eines Arzneimittels wird an einer relativ geringen Zahl von Patienten durchgeführt (in der Regel mehrere 1000 Patienten für eine Phase-III-Studie). Diese Patienten sind zudem durch Ein- und Ausschlusskriterien stratifiziert (siehe Teil 2) und bilden somit nicht das gesamte Patientenkollektiv ab, das für eine bestimmte Indikation infrage kommt.
Seltene Nebenwirkungen, Arzneimittelinteraktionen, Interaktionen mit Lebensmitteln usw. können in klinischen Prüfungen nicht abschließend untersucht werden. Tabelle 1 gibt einen Überblick, wie viele Patienten statistisch benötigt werden, um Nebenwirkungen zu entdecken.
Tab. 1: Erforderliche Anzahl von Patienten, um mit 95%iger Wahrscheinlichkeit eine Arzneimittelnebenwirkung zu detektieren. Seltene und sehr seltene Nebenwirkungen mit einer Inzidenz von 1:10.000 oder 1:100.000 werden in klinischen Studien mit maximal 3000 Patienten nicht erfasst (grüne Linie). Nach [5]. | |
Häufigkeit der Nebenwirkung |
Patienten (n) |
1: 10 (sehr häufig) |
30 |
1: 100 (häufig) |
300 |
1: 1.000 (gelegentlich) |
3.000 |
1: 10.000 (selten) |
30.000 |
1: 100.000 (sehr selten) |
300.000 |
Aufgabe der Pharmakovigilanz
Neue Sicherheitsaspekte können sich noch Jahre nach der Zulassung ergeben und hängen von neuen Entwicklungen in der medizinischen Wissenschaft ab. Das heißt, sie müssen über einen stetigen Informationsfluss aktualisiert werden. Das Arzneimittelgesetz (AMG) und auch die europäische Gesetzgebung sehen vor, dass nach der Zulassung eines Arzneimittels die Erfahrungen bei seiner Anwendung fortlaufend systematisch gesammelt werden und gegebenenfalls Maßnahmen ergriffen werden; dies sind die Hauptaufgaben der Pharmakovigilanz (griech. pharmakon = Arzneimittel; lat. vigilare = wachsam sein).
Die Pharmakovigilanz etablierte sich in Deutschland im Zusammenhang mit der Contergantragödie 1959 – 61, als tausende missgebildete Kinder zur Welt kamen (siehe Teil 1). Als Konsequenz daraus verabschiedete die WHO 1963 eine Resolution zur schnellen Reaktion und Informationsweiterleitung bei schwerwiegenden Nebenwirkungen und gründete 1968 ein Pilotforschungsprogramm, das ein internationales System zur Detektion von Nebenwirkungen entwickeln sollte. Der technische Report, der 1971 als Ergebnis daraus hervorging, diente als Grundlage zur Etablierung eines länderübergreifenden Pharmakovigilanzsystems, das seitdem stetig weiterentwickelt und in die Gesetzgebung der Mitgliedsländer integriert wurde. Dies geschah in der Bundesrepublik durch das Gesetz zur Neuordnung des Arzneimittelrechts von 1976 (AMG 1976). Damals wurde erstmals ein Pharmakovigilanzsystem implementiert, das in seinen Grundzügen auch heute noch gültig ist.
Definition: PharmakovigilanzPharmakovigilanz ist laut Definition der Weltgesundheitsorganisation WHO die "Wissenschaft und alle Aktivitäten, die zur Entdeckung, Beurteilung sowie zum Verständnis und zur Vorbeugung von unerwünschten Wirkungen oder anderen Problemen in Verbindung mit Arzneimitteln dienen". |
Das Spontanmeldesystem
Das Beobachten und Sammeln von Verdachtsfällen unerwünschter Arzneimittelwirkungen (UAW, siehe Infobox) nach der Zulassung ist eine wichtige Aufgabe der Pharmakovigilanz.
INFOBOX
Nebenwirkung und unerwünschte Arzneimittelwirkung (UAW)Im deutschen Sprachgebrauch und auch im Arzneimittelgesetz (AMG) wird der Begriff der Nebenwirkung für unerwünschte Arzneimittelwirkung(en) (UAW) verwendet. Im eigentlichen Wortsinn bedeutet Nebenwirkung jeglichen Effekt eines Arzneimittels, der von der Hauptwirkung abweicht, unabhängig davon, ob er erwünscht oder unerwünscht ist. Aus diesem Grunde wird im AMG nach § 4 Abs. 13 der Begriff der Nebenwirkung konkreter definiert (16. AMG-Novelle): "Nebenwirkungen sind … schädliche, unbeabsichtigte Reaktionen auf das Arzneimittel …" Danach umfassen Nebenwirkungen bei Humanarzneimitteln nicht nur solche, die beim bestimmungsgemäßen Gebrauch auftreten (Definition vor der 16. AMG-Novelle), sondern auch Reaktionen infolge von Überdosierung, Fehlgebrauch, Missbrauch und Medikationsfehlern sowie Nebenwirkungen, die mit beruflicher Exposition verbunden sind. Im englischen Sprachgebrauch findet der treffendere Ausdruck adverse drug reactions (unerwünschte Arzneimittelwirkungen, UAW) Anwendung, der alle unbeabsichtigten schädlichen Effekte bezeichnet. Des Weiteren werden im § 4 Abs. 13 AMG noch schwerwiegende und unerwartete Nebenwirkungen definiert (siehe Teil 2). |
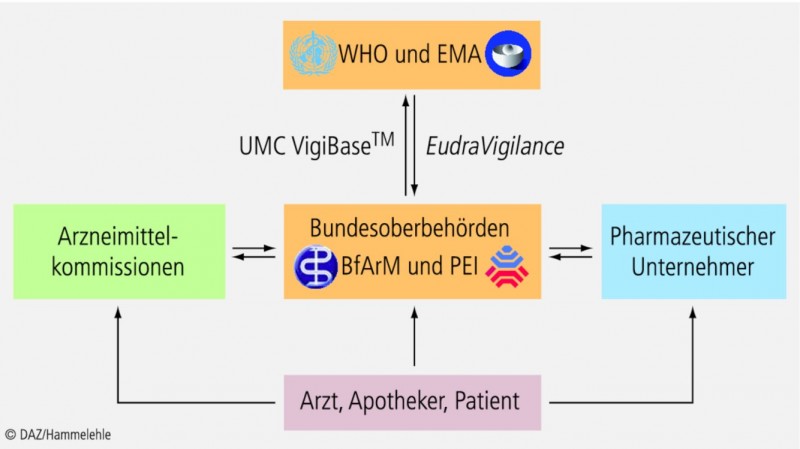
Das Frühwarnsystem zur Detektion von neuen arzneimittelbedingten Risiken beruht auf dem Sammeln und Auswerten von Spontanberichten über UAW, die von Ärzten, Zahn- und Tierärzten, Apothekern und Patienten stammen. Eine neue Information über ein mögliches Risiko, die Anlass zur weiteren Abklärung gibt, bezeichnet man in diesem Zusammenhang auch als "Signal". Von besonderer Bedeutung sind dabei schwerwiegende und bis dato unbekannte UAW. Ärzte und Apotheker sind über ihre Berufsordnungen dazu verpflichtet, UAW zu melden. Dabei können sie sich
- an den pharmazeutischen Unternehmer des betreffenden Medikamentes oder
- an die zuständigen Bundesoberbehörden (BfArM oder PEI) oder
an die Arzneimittelkommissionen der Heilberufskammern (z. B. die Arzneimittelkommission der Deutschen Apotheker, AMK, oder die Arzneimittelkommission der deutschen Ärzteschaft, AkdÄ)
wenden. Die pharmazeutischen Unternehmer und die Arzneimittelkommissionen sind gesetzlich dazu verpflichtet, alle gemeldeten Verdachtsfälle an die Bundesoberbehörden weiterzuleiten, wo sie gesammelt und ausgewertet werden (Abb. 1).
Verdachtsfälle schwerwiegender UAW von Arzneimitteln muss der pharmazeutische Unternehmer innerhalb von 15 Tagen elektronisch in einem standardisierten Format an die Bundesoberbehörde übermitteln. Bei nicht schwerwiegenden UAW reicht eine Einzelfallmeldung innerhalb von 90 Tagen. Bei der Meldung von UAW ist eine international standardisierte medizinische Terminologie nach dem Medical Dictionary for Regulatory Activities (MedDRA) zu verwenden, die von der International Conference on Harmonization (ICH, siehe Teil 1) verabschiedet und verbindlich in EU-Recht implementiert wurde.
Die einheitliche Terminologie ist von großer Wichtigkeit, da die Behörden aller EU-Staaten ihre Meldungen in die zentrale europäische Datenbank EudraVigilance bei der Europäischen Arzneimittelagentur (EMA) einspeisen müssen. Zusätzlich werden die Meldungen auch an die globale Datenbank VigiBase der WHO beim Uppsala Monitoring Centre (UMC) in Schweden weitergeleitet.
Die kürzlich in Kraft getretene Pharmakovigilanzgesetzgebung (16. AMG-Novelle) sieht vor, dass Patienten über einen Standardtext in der Packungsbeilage dazu aufgefordert werden, UAW an den Arzt, Apotheker oder unmittelbar an die Bundesoberbehörde zu melden. Ebenfalls neu ist, dass auch Ärzte und andere Gesundheitsberufe in den Fachinformationen eines Arzneimittels (siehe Infobox) dazu aufgefordert werden, Verdachtsfälle einer UAW an die Behörde zu melden. Seit 2012 besteht auch die Möglichkeit für Patienten, auf der Homepage www.adrreports.eu Einsicht in die europäische Meldedatenbank für Verdachtsfälle von UAW (EudraVigilance) zu bekommen.
INFOBOX
FachinformationDie Fachinformation ist die Basisinformation eines Arzneimittels für die Heilberufe. Darin finden sich alle relevanten Informationen für einen sicheren und effektiven Gebrauch des Arzneimittels. Der Inhalt der Gebrauchsinformation (Packungsbeilage) für den Anwender leitet sich aus den Informationen der Fachinformation ab. Folgende Daten sind u. a. in der Fachinformation zu finden:
|
Die Vorteile dieses Spontanmeldesystems liegen darin, dass es vergleichsweise günstig ist und dass – anders als bei klinischen Prüfungen – Informationen über ein Arzneimittel unter Alltagsbedingungen gewonnen werden. Zum Beispiel können UAW gemeldet werden, die aus dem Gebrauch des Arzneimittels an Kindern und älteren Menschen stammen, oder auch Wechselwirkungen mit anderen Medikamenten oder Nahrungsmitteln, die vorher nicht Gegenstand der klinischen Prüfung waren.
Nachteile dieses Systems sind die geringe Anzahl der tatsächlich gemeldeten Fälle und die oftmals schlechte Qualität und Unvollständigkeit der Meldungen. Schätzungsweise werden nur zwischen 5 bis 10% der Verdachtsfälle auf UAW tatsächlich gemeldet (engl. underreporting). Dieser Anteil ist relativ gering, wenn man bedenkt, dass die vorgegebene Schwelle zur Meldung solcher Verdachtsfälle sehr niedrig ist. So sollten schon bloße Vermutungen, dass eine beobachtete UAW mit einem Arzneimittel im Zusammenhang steht, gemeldet werden; dies unterbleibt jedoch häufig.
Ein weiterer Nachteil ist die geringe Meldung von Verdachtsfällen auf UAW beim Gebrauch des Arzneimittels außerhalb seiner Zulassung, dem sogenannten Off-label-use. Er kommt häufig in der Kinder- und Jugendmedizin vor, weil viele Medikamente für diese Altersgruppe nicht zugelassen sind. Hier ist die Hemmschwelle der verordnenden Ärzte häufig besonders hoch, Meldungen abzugeben.
Der Periodic Safety Update Report – PSUR
Zu den Dokumentations- und Meldepflichten, denen der pharmazeutische Unternehmer unterliegt, gehören neben den Spontanmeldungen auch regelmäßige aktualisierte Berichte über die Unbedenklichkeit des Arzneimittels – Periodic Safety Update Report (PSUR) – an die Bundesoberbehörde (§ 63 b AMG). Die Erstellung von PSURs ist für alle in der EU, den USA und in Japan in den Verkehr gebrachten Arzneimittel obligatorisch (also in den ICH-Mitgliedsländern, siehe Teil 1).
Der Zweck des PSUR liegt darin, eine weltweite Nutzen-Risiko-Bewertung eines Arzneistoffs vorzunehmen. Im Gegensatz zur Bewertung einer einzelnen UAW-Meldung, mit der häufig keine definitive Aussage zur Veränderung des Nutzen-Risiko-Profils eines Arzneimittels gemacht werden kann, ermöglicht es der PSUR, eine fundierte Risikoanalyse des betroffenen Arzneistoffes durchzuführen. Für den pharmazeutischen Unternehmer bedeutet dies, dass er nicht nur die vorhandenen Informationen über "sein" Medikament bereitstellen muss, sondern alle weltweiten, sicherheitsrelevanten Fakten zum jeweiligen Arzneistoff. Dabei spielt es keine Rolle, ob der Wirkstoff auch von einer anderen Firma vermarktet wird. PSURs müssen in definierten Zeiträumen eingereicht werden:
- nach der Zulassung bis zum ersten Inverkehrbringen alle sechs Monate,
- nach dem ersten Inverkehrbringen sechsmonatlich in den ersten beiden Jahren, jährlich in den zwei folgenden Jahren, danach alle drei Jahre.
Um den Verwaltungsaufwand zu verringern, haben pharmazeutische Unternehmer über eine Ausnahmeregelung die Möglichkeit, gemeinschaftliche PSURs zu einem Arzneistoff zu bestimmten Stichtagen einzureichen.
Innerhalb Europas weisen Fachinformationen zu Arzneimitteln des Öfteren Unterschiede auf. Um diese zu harmonisieren, muss der Innovator eines Arzneistoffs ein sogenanntes Core Safety Profile (CSP) vorschlagen, in dem alle für die Fachinformation relevanten Sicherheitsaspekte des Arzneistoffs aufgeführt sind. Dieses CSP wird dann im Rahmen eines sogenannten PSUR Worksharing-Prozesses zwischen den EU-Mitgliedsländern ergänzt und verabschiedet. Ein verabschiedetes CSP dient als Referenzdokument für alle anderen Arzneimittel mit demselben Arzneistoff. Sollten sich durch einen späteren PSUR sicherheitsrelevante Änderungen in der Fachinformation ergeben, müssen diese im CSP aktualisiert werden.
Der Risk Management Plan – RMP
Neben dem PSUR besteht für den Zulassungsinhaber eines Arzneimittels die Verpflichtung, einen Risk Management Plan (RMP) zu erstellen. Dieses Dokument komplementiert die im PSUR vorgenommene Nutzen-Risiko-Bewertung durch die Beschreibung eines Systems zum Nutzen-Risiko-Management. Durch dieses Nutzen-Risiko-Management soll sichergestellt werden, dass der Nutzen eines Arzneimittels zu jeder Zeit die Risiken um das größtmögliche Maß überschreitet. Der RMP enthält dazu eine umfassende Beschreibung des Sicherheitsprofils des jeweiligen Arzneistoffs. Von besonderer Bedeutung sind hierbei Risiken, die die Unbedenklichkeit eines Arzneimittels nachhaltig beeinflussen könnten, sogenannte wichtige Risiken. Darauf aufbauend muss der RMP einen strukturierten Plan enthalten, in dem der Unternehmer angibt, wie er
- neue Risiken identifiziert,
- bekannte Risiken charakterisiert und deren Risikofaktoren aufklärt,
- untersucht, ob potenzielle Risiken real sind oder nicht,
- fehlende Informationen sammelt,
plant, identifizierte, potenzielle und auch unbekannte Risiken (z. B. aufgrund des Ausschlusses von bestimmten Patientengruppen in klinischen Studien) zu reduzieren, zu vermeiden und zu minimieren.
Letzteres kann beispielsweise durch Erwähnung in der Fach- oder Gebrauchsinformation, Beschränkung der Packungsgröße, Verschreibungspflicht, Patientenregister oder auch Studien zur Arzneimittelsicherheit gewährleistet werden.
Studien zur Arzneimittelsicherheit
Wenn das Medikament auf dem Markt ist und routinemäßig eingesetzt wird, werden weiterhin gezielt Daten zu Nebenwirkungen oder Interaktionen gesammelt. Im 2. Teil dieser Serie wurde in diesem Zusammenhang bereits die Anwendungsbeobachtung (siehe Phase-IV-Studien) als Instrument der Pharmakovigilanz angesprochen.
Bestehen bereits bei der Zulassung Bedenken bezüglich Sicherheit oder Wirksamkeit eines Arzneimittels, kann dem pharmazeutischen Unternehmer zur Auflage gemacht werden, für die Anfangszeit nach der Zulassung weitere Studien durchzuführen. Je nachdem, was in den Studien untersucht werden soll, nennt man diese auch Post Authorisation Safety Studies (PASS) oder Post Authorisation Efficacy Studies (PAES). Solche Studien können auch im Rahmen von auftretenden Sicherheitsbedenken eines Arzneimittels nachträglich von der zuständigen Bundesoberbehörde als Auflage angeordnet werden (s. u. "Stufenplanverfahren").
Medikamente, die einer zusätzlichen Überwachung bedürfen, werden von der EMA in einer Liste geführt. Sie sollen zukünftig zur Erhöhung der Aufmerksamkeit durch ein schwarzes Symbol in der Packungsbeilage oder Fachinformation gekennzeichnet werden.
Die Daten aus Spontanmeldungen, Anwendungsbeobachtungen, PASS/PAES usw. werden in epidemiologischen Datenbanken gespeichert. Mithilfe solcher Datenbanken lassen sich neu eingeführte Arzneimittel weitläufig überwachen, Häufigkeiten auftretender UAW abschätzen und regulatorische Maßnahmen evaluieren.
Das Stufenplanverfahren
Kommt die Bundesoberbehörde nach der Auswertung von UAW-Meldungen zu dem Schluss, dass ein Verdacht auf ein gesundheitliches Risiko für die Bevölkerung besteht, kann sie das sogenannte Stufenplanverfahren auslösen. Nach § 63 AMG handelt es sich beim Stufenplan um eine "Allgemeine Verwaltungsvorschrift zur Beobachtung, Sammlung und Auswertung von Arzneimittelrisiken". Der Begriff Stufenplan leitet sich von der Unterteilung des Verfahrens in zwei Gefahrenstufen ab (Überblick in Abb. 2).
Die Stufe I des Verfahrens beginnt, wenn es Hinweise auf ein mögliches Arzneimittelrisiko gibt. Dabei reicht bereits ein assoziationsartiger Bezug zwischen der Beobachtung einer UAW und der Gabe eines Arzneimittels aus, z. B. eine bei bestimmungsgemäßem Gebrauch auftretende UAW oder Wechselwirkungen mit anderen Mitteln. Aber auch auf den ersten Blick banalere Mängel an Packungsbeilage, Behältnissen und äußeren Umhüllungen können zur Einleitung eines Stufe-I-Verfahrens führen. Zur Klärung des Sachverhaltes findet zunächst ein Informationsaustausch zwischen der Bundesoberbehörde, dem pharmazeutischen Unternehmer und anderen im Stufenplan festgelegten Behörden und Stellen statt. Insgesamt werden im Stufenplan 14 solcher Behörden und Stellen aufgeführt, mit denen die Bundesoberbehörden ggfs. zusammenwirken, z. B. die obersten Landesgesundheitsbehörden, die Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (ZLG; https://www.zlg.de) oder die Arzneimittelkommissionen der Heilberufskammern.
Der pharmazeutische Unternehmer hat die Verantwortung, dem derzeitigen wissenschaftlichen Erkenntnisstand entsprechende Produkte auf den Markt zu bringen. Somit obliegt es ihm auch, eigenverantwortlich adäquate Maßnahmen wie z. B. eine Änderung der Fachinformation vorzunehmen. Kommt die Bundesoberbehörde nach Prüfung aller Unterlagen und der vorgeschlagenen Maßnahmen zu der Erkenntnis, dass kein Risiko mehr besteht, wird das Verfahren nach der Stufe I beendet. Sollte sich aber ein begründeter Verdacht auf Arzneimittelrisiken herauskristallisieren und der pharmazeutische Unternehmer nicht dazu in der Lage sein, angemessene Maßnahmen zu treffen, wird ein Stufe-II-Verfahren eingeleitet. Auch kann bei einem von Anfang an begründeten Verdacht auf Arzneimittelrisiken direkt ein Stufe-II-Verfahren eingeleitet werden.
Meist kommt es in der Stufe II nur zu schriftlichen Anhörungen des pharmazeutischen Unternehmers über die geplanten Maßnahmen. Nach Begutachtung und gegebenenfalls Modifizierung führt er in den meisten Fällen die von der Bundesoberbehörde erforderlich gehaltenen Maßnahmen aus.
Die anzuordnenden Maßnahmen fallen, je nach Art der Maßnahme, in unterschiedliche Zuständigkeitsbereiche. So ist das Bundesministerium für Gesundheit zuständig für arzneistoffbezogene Maßnahmen von Arzneimitteln, z. B. die Ausweitung der Apothekenpflicht (§ 46 AMG), die Unterstellung unter die Verschreibungspflicht (§ 48 AMG) oder die Unterstellung unter das Betäubungsmittelrecht.
Die Landesgesundheitsbehörden sind zuständig, wenn es z. B. darum geht, bestimmte Chargen eines Arzneimittels aus dem Verkehr zu nehmen ("Anordnung des Rückrufes" nach § 69 Abs. 1 AMG).
Die Bundesoberbehörde kann in letzter Instanz z. B. die Zulassung eines Arzneimittels widerrufen, sie zurücknehmen oder ruhen lassen.
Häufig können Arzneimittelrisiken bereits durch Auflagen an den pharmazeutischen Unternehmer bei der Marktzulassung oder später bei einer Änderungsanzeige (s. u.) verringert werden. Zu diesen gehören u. a.:
- Ergänzungen des Wortlautes der Packungsbeilage oder Fachinformation
- Änderungen oder Ergänzungen des Wortlautes auf der Verpackung
- Anordnung einer therapiegerechten Packungsgröße
- Aufnahme von Warnhinweisen in die Packungsbeilage oder Fachinformation
Anordnung einer systematischen Sammlung und Dokumentation von Erkenntnissen nach der Zulassung (Phase IV)
Anordnung, in welcher Form der pharmazeutische Unternehmer Änderungen der Fachinformation den Fachkreisen bekannt zu geben hat (z. B. durch "Rote-Hand-Briefe", siehe Infobox mit Abb. 3)
- Anordnung von PASS/PAES (s. o.)
- Anordnung des Sofortvollzuges.
INFOBOX
Rote-Hand-Briefe
Im Rahmen der Verpflichtung des pharmazeutischen Unternehmers, eigenverantwortlich Maßnahmen zur Vermeidung von Gesundheitsschäden zu ergreifen, kann er eine "wichtige Mitteilung über ein Arzneimittel" als Rundbrief mit dem Symbol einer Roten Hand versenden. Zielgruppe der "Rote-Hand-Briefe" sind vor allem Ärzte, Apotheker und Verbraucher. Das Schreiben besitzt ein definiertes Layout und soll bereits in der Überschrift auf Kernpunkte hinweisen und die wichtigen Informationen über neue Arzneimittelrisiken frei von Werbung in Kürze darlegen. Form und Aufmachung des Briefes mit der Roten Hand sind so gehalten, dass er direkte "Aufmerksamkeit" erregt.
Den Rote-Hand-Brief kann man auch als E-Mail abonnieren, beispielsweise bei der AkdÄ.
Der pharmazeutische Unternehmer hat das Recht, Widerspruch gegen angeordnete Maßnahmen einzulegen (Rechtsmittel), und kann somit die Umsetzung der Maßnahmen aufschieben (Suspensiveffekt). Allerdings kann der Sofortvollzug (s. o.) angeordnet werden, also die sofortige Umsetzung der angeordneten Maßnahmen, wenn ein begründeter Verdacht auf Bedenklichkeit eines Arzneimittels besteht ("Gefahr im Verzug"). Dies kann z. B. der Fall sein, wenn eine Marktrücknahme angeordnet wurde oder von schweren Risiken auszugehen ist ("ungünstiges Nutzen-Risiko-Verhältnis"), die es unvertretbar machen, den Abschluss des Rechtsmittelverfahrens abzuwarten. Einen besonderen Status haben hierbei biologische oder biotechnologische Arzneimittel. Der Gesetzgeber geht bei ihnen per se von einer stärkeren Gefährdung der Bevölkerung bei auftretenden Qualitätsmängeln aus. Aus diesem Grunde ist der gesetzliche Sofortvollzug bei ihnen für bestimmte Auflagen zu Herstellung und Kontrolle im AMG vorgegeben (§ 28 Abs. 3 c Satz 2).
Wenn ein Gesundheitsrisiko für die Bevölkerung besteht (engl. risk to public health) und schnelles Handeln erforderlich ist, können sowohl der pharmazeutische Unternehmer als auch die Bundesoberbehörde oder die EMA eine "Dringende Zulassungsänderung" (engl. urgent safety restrictions, USR) einleiten bzw. anordnen. Die Änderungen beziehen sich auf die Produktinformation des Arzneimittels z. B. in den Abschnitten Indikation, Dosierung, Gegenanzeigen und Warnhinweise (siehe auch Infobox Fachinformation). Der pharmazeutische Unternehmer muss die Bundesoberbehörde und/oder die EMA mindestens 24 Stunden vorher über die geplanten USR in Kenntnis setzen. Sollten nach Ablauf der 24-Stunden-Frist keine Einwände erhoben worden sein, gilt die USR als akzeptiert und kann ausgeführt werden. Allerdings muss der pharmazeutische Unternehmer innerhalb von 15 Tagen noch einmal einen offiziellen Antrag auf Änderung der Fachinformation in Form einer Änderungsanzeige/Variation (s. u.) einreichen.
Ärzte und Apotheker werden von der Bundesoberbehörde nicht direkt über die Maßnahmen informiert. Die Zuständigkeit hierfür liegt bei den Kammern der Heilberufe, die entsprechende Mitteilungen in der Fachpresse machen. Des Weiteren kann der pharmazeutische Unternehmer Ärzte und Apotheker auch direkt durch "Rote-Hand-Briefe" über eine USR informieren (siehe Infobox).
INFOBOX
Beispiel einer "Dringenden Zulassungsänderung" (USR)Im August 2007 kam es in Deutschland und der EU durch eine USR zu Anwendungsbeschränkungen des Arzneimittels Prexige®. Dessen Wirkstoff Lumiracoxib gehört zur Klasse der selektiven COX-II-Inhibitoren oder Coxibe, die zur Behandlung von Arthrose und akuten Schmerzen eingesetzt werden. Lumiracoxib führte vor allem in hohen Dosen gehäuft zu schwerwiegenden Leberschädigungen der Patienten. Danach erfolgte in Australien die Marktrücknahme des Medikaments und in der EU zunächst eine USR. Mit einem Rote-Hand-Brief informierte der Hersteller die Fachkreise, dass ab sofort vor Beginn der Behandlung sowie regelmäßig während der Behandlung Kontrollen der Leberenzymwerte vorgeschrieben werden. Weiterhin sollte Lumiracoxib nicht mehr bei Patienten mit erhöhtem Risiko für Leberschädigungen angewendet werden. In den folgenden drei Monaten stellte sich heraus, dass auch in niedrigeren Dosen und bereits nach vergleichsweise kurzer Anwendungsdauer Leberschädigungen auftraten. Die vorherige USR war von einer anderen Datenlage ausgegangen und reichte daher nicht aus, das Risiko zu mindern. Als Konsequenz ordnete das BfArM im Rahmen eines Stufenplanverfahrens der Stufe II das Ruhen der Zulassung für alle Lumiracoxib-haltigen Arzneimittel an. Diese Entscheidung wurde erneut vom Hersteller in einem Rote-Hand-Brief an die Fachkreise kommuniziert. |
Mit der 2. AMG-Novelle (1986) wurde die Funktion des Stufenplanbeauftragten geschaffen. Jeder pharmazeutische Unternehmer, der Fertigarzneimittel in den Verkehr bringt, muss mit dieser Funktion eine in der EU ansässige, qualifizierte Person (qualified person) beauftragen. Ausgenommen sind von dieser Regelung u. a. Apothekeninhaber, die Arzneimittel für den üblichen Apothekenbetrieb herstellen, oder Tierärzte. Die im AMG und in der Betriebsverordnung für pharmazeutische Unternehmer (PharmBetrV) festgelegten Aufgaben des Stufenplanbeauftragten kann man so zusammenfassen: Sammlung aller bekannt gewordenen Meldungen über Arzneimittelrisiken, deren Bewertung und Dokumentation sowie die eventuell notwendige Koordination von Maßnahmen.
Der Stufenplanbeauftragte steht dabei auch in der Pflicht, der Bundesoberbehörde (schwerwiegende) UAW innerhalb der geforderten Fristen (s. o.) anzuzeigen. Bei Versäumung der Fristen, falschen oder unvollständigen UAW-Meldungen macht er sich strafbar. Weiterhin muss er auf Verlangen der zuständigen Bundesoberbehörde weitere Informationen für die Beurteilung des Nutzen-Risiko-Verhältnisses eines Arzneimittels, einschließlich eigener Bewertungen, unverzüglich und vollständig übermitteln.
Das Risikoverfahren der Europäischen Union
Durch die Umsetzung der europäischen Gesetzgebung in nationales Recht verschieben sich die Kompetenzen immer mehr von den nationalen Zulassungsbehörden zur EMA und dem neugeschaffenen Ausschuss für Risikobewertung (Pharmacovigilance Risk Assessment Committee, PRAC). So wird ein europäisches Referral-Verfahren (siehe Infobox) ausgelöst, wenn aufgrund von UAW-Meldungen eine Änderung, ein Ruhen oder ein Widerruf der Zulassung in Erwägung gezogen wird. Nur bei national zugelassenen Arzneimitteln, die allein in Deutschland auf dem Markt sind, kommt auch weiterhin das nationale Stufenplanverfahren zur Anwendung.
INFOBOX
Das europäische Referral-VerfahrenWenn zwischen den Mitgliedstaaten der EU Unstimmigkeit über ein bestimmtes Arzneimittel oder eine Wirkstoffklasse herrscht, können sie in einem Referral (wörtlich: Rückübertragung) die EMA um eine wissenschaftliche Bewertung bitten. Die EMA beauftragt den CHMP mit der Durchführung des Referrals und veröffentlicht die Position des CHMP (CHMP-Opinion) nach Abschluss des Referrals auf der EMA-Homepage. Es gibt eine ganze Reihe von Gründen, ein Referral zu starten. Sie reichen von Bedenken über die Arzneimittelsicherheit bis zu Meinungsverschiedenheiten zwischen den Mitgliedstaaten über die Anwendungsgebiete, Dosierung oder Kontraindikationen des Arzneimittels. Das Recht, ein Referral zu initiieren, haben die Mitgliedsländer (durch ihre nationalen Behörden) und der pharmazeutische Unternehmer. |
Das BfArM, das PEI und die EU-Kommission sind berechtigt, bei bedenklichen Pharmakovigilanzdaten ein Dringlichkeitsverfahren einzuleiten (Artikel 107-Verfahren, Urgent Union Procedure). Im Rahmen dieses Verfahrens gibt der PRAC Empfehlungen an den Ausschuss für Humanarzneimittel (CHMP) oder die sogenannte Koordinierungsgruppe (CMDh, Coordination Group for Mutual Recognition and Decentralized Procedures, human), die auf dieser Grundlage Gutachten zur Entscheidungsfindung erstellen.
Je nachdem, welche Gremien (Koordinierungsgruppen oder die EU-Kommission) an einer Entscheidung beteiligt waren, hat die Entscheidung bindenden oder empfehlenden Charakter. Ihre Umsetzung erfolgt weiterhin über ein nationales Stufenplanverfahren. Zur Erhöhung der Transparenz gibt die EMA die Einleitung eines Dringlichkeitsverfahrens auf ihrem Internetportal bekannt (www.ema.europa.eu/ema).
Änderungsanzeigen
Änderungsanzeigen (engl. variations) betreffen Änderungen der Zulassung von Arzneimitteln und dabei hauptsächlich deren Fach- und Gebrauchsinformation. Im Hinblick auf eventuelle Änderungen der Unbedenklichkeit des Arzneimittels werden sie in vier Kategorien eingeteilt:
1. Änderung des Typs IA:
Sie müssen der Behörde nur angezeigt werden und dienen der schnellen Umsetzung von kleineren Änderungen (engl. minor changes), wie beispielsweise der Änderung der Kontaktdaten des Zulassungsinhabers. Die Einstufung einer Änderungsanzeige als Typ IA erfolgt anhand einer speziellen Liste.
2. Änderung des Typs IB: Hierzu zählen ebenfalls minor changes, die allerdings nicht von der oben genannten Liste abgedeckt sind, aber auch nicht als größere Änderungen einzustufen sind (Typ-II-Änderungen, s. u.). Im Gegensatz zu Typ-IA-Änderungen können Änderungen des Typs IB von der zuständigen Behörde innerhalb einer Frist von 30 Tagen abgelehnt werden.
3. Änderung des Typs II:
Es sind größere Änderungen (major changes) wie beispielsweise das Hinzufügen einer neuen therapeutischen Indikation. Vor ihrer Umsetzung ist die sogenannte "Vorabgenehmigung" durch die zuständige Behörde erforderlich.
4. Zulassungserweiterung: Eine Zulassungserweiterung (engl. extension) ist bei bestimmten Änderungen eines Arzneimittels erforderlich, die in einer weiteren Liste stehen. Darunter fallen beispielsweise Änderungen der Formulierung (z. B. durch Einsatz eines anderen Salzes oder Esters). Diese dürfen jedoch keinen signifikanten Einfluss auf die Wirksamkeit und Unbedenklichkeit des Arzneimittels haben. Ähnlich wie bei Typ-II-Änderungsanzeigen darf eine Erweiterung nur durchgeführt werden, nachdem die maßgebliche Behörde über den Antrag entschieden hat.
Verlängerung der Zulassung
Bis 2005 mussten pharmazeutische Unternehmer alle fünf Jahre eine Verlängerung der Zulassung ihres Arzneimittels bei der Bundesoberbehörde beantragen. Seit der 14. AMG-Novelle von 2005 muss er nur noch einmal einen Antrag auf Verlängerung (engl. renewal) stellen, nämlich fünf Jahre nach der Erstzulassung. Hiernach gilt die Zulassung in der Regel unbefristet.
Wird allerdings ein Arzneimittel nach erteilter Zulassung nicht vermarktet, so erlischt seine Zulassung bereits nach drei Jahren (§ 31 AMG). Diese als "Sunset Clause" bekannte Regelung gründet sich ebenfalls auf die 14. AMG-Novelle. Der Zulassungsinhaber ist dazu verpflichtet, der zuständigen Bundesoberbehörde das Datum des Inverkehrbringens eines zugelassenen Arzneimittels unter Berücksichtigung der unterschiedlichen zugelassenen Darreichungsformen und Wirkstärken unverzüglich anzuzeigen (§ 29 Abs. 1 b AMG). Zudem muss er der zuständigen Bundesoberbehörde das Datum der Einstellung des Inverkehrbringens mitteilen.
Die Abbildung 4 fasst das Prozessmanagement des gesamten Lebenszyklus eines Arzneimittels von der Entwicklung des Wirkstoffes über die präklinischen und klinischen Prüfungen bis zur Zulassung und Markteinführung des Arzneimittels und die sich daran anschließende Beobachtung seiner Anwendung unter Alltagsbedingungen zusammen.
Literatur
[1] Deutsches Arzneimittelgesetz (AMG) in der gültigen Fassung; www. gesetze-im-internet.de/amg_1976.
[2] Fuhrmann S, Klein B, Fleischfresser A. Arzneimittelrecht: Handbuch für die pharmazeutische Rechtspraxis. Baden-Baden 2010.
[3] Einführung in die Grundlagen der Pharmakovigilanz (Teile I – III). Bulletin zur Arzneimittelsicherheit – Informationen aus BfArM und PEI 01/2010:14 – 17; 04/2010:18 – 24; 02/2011:17 – 21.
[4] Farzan J. Neue Pharmakovigilanz-Gesetzgebung in der EU. Bulletin zur Arzneimittelsicherheit – Informationen aus BfArM und PEI 03/2012: 14 – 17.
[5] Porta MS, Hartzema AG. The contribution of epidemiology to the study of drugs. Drug Intell Clin Pharm 1987;21:741 – 747.
Autoren
Dr. Bodo Haas*, bodohaas@web.de
Dr. Nils Lilienthal, Nils.Lilienthal@arcor.de
Dr. Niels Eckstein, Niels.Eckstein@web.de
* korrespondierender Autor



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.