














- DAZ.online
- DAZ / AZ
- DAZ 29/2012
- Vom Wirkstoff zum ...
UniDAZ
Vom Wirkstoff zum Arzneimittel
EditorialLiebe Leserinnen, liebe Leser!In der DAZ Nr. 25 begann die Serie "Vom Wirkstoff zum Arzneimittel" mit der Darstellung der präklinischen, experimentellen Studien, die an Zellkulturen und Labortieren durchgeführt werden. Hier im 2. Teil der Serie erfahren Sie alles Wichtige über die klinischen Studien, das heißt: die Testung von Wirkstoffen am Menschen: zuerst an gesunden Probanden, dann an Patienten. Im dritten Teil wird es darum gehen, wie man das in den Markt eingeführte Arzneimittel weiter beobachtet und unter Alltagsbedingungen Erfahrungen sammelt. Wir laden Sie wieder ein, selbst aktiv an UniDAZ mitzuwirken. Surfen Sie auf unsere Homepage www.unidaz.de, wo Sie bei den Hochschulporträts und den Blogs sogar zum Autor werden können. Zu Beginn des Wintersemesters erscheint wieder ein UniDAZ-Magazin. Wenn Sie Ideen, Kritik oder Vorschläge haben, schreiben Sie uns eine Mail an redaktion@unidaz.de. Und wenn Sie ein UniDAZ-Fan sind, klicken Sie auf den "Gefällt mir"-Button der UniDAZ-Facebook-Seite. Ihre UniDAZ-Redaktion |
Die Gesetze, Verordnungen und vor allem Leitlinien machen Vorgaben, wie eine klinische Prüfung durchzuführen ist. Es müssen unter anderem Studienprotokolle erstellt werden, die Studiendesigns beschrieben werden und die "Good Clinical Practice” (GCP)-Leitlinie beachtet werden (s. u.).
Ethische Aspekte werden berücksichtigt, indem die klinische Prüfung entsprechend der Deklaration von Helsinki (siehe Infobox) durchgeführt wird. In jeder der vier Phasen der klinischen Prüfung eines Medikamentes werden die Ziele der Prüfung, die Testpersonen, die Behandlungsdauer und die Studiendesigns angepasst.
Definition: Klinische PrüfungDer Begriff Klinische Prüfung ist in § 4 Abs. 23 AMG wie folgt definiert: "Eine klinische Prüfung bei Menschen ist: jede am Menschen durchgeführte Untersuchung, die dazu bestimmt ist,
mit dem Ziel, sich von der Unbedenklichkeit oder Wirksamkeit der Arzneimittel zu überzeugen." |
Infobox: Ethik-KommissionDie Ethikkommission (EK) ist der unabhängige Sachwalter der Patientenund Probandenrechte. Ethikkommissionen sind entweder an den medizinischen Fakultäten von Universitäten oder bei den Landesärztekammern angesiedelt. Zurzeit gibt es 53 Ethikkommissionen in Deutschland. Jeder Antrag auf Durchführung einer klinischen Studie muss vor Beginn der Rekrutierung zusätzlich zur behördlichen Genehmigung ein positives Votum der zuständigen EK einholen. Die EK ist ein Gremium, dem neben (medizinischen und pharmazeutischen) Experten und Juristen auch medizinische Laien, z. B. Philosophen und Theologen angehören können. Typischerweise handelt es sich um Hochschullehrer. Der EK kommen zwei wichtige Funktionen zu: Zum einen beurteilt sie die medizinische Forschung am Menschen rechtlich und ethisch, zum anderen berät sie die Ärzte als Kammermitglieder in berufsrechtlichen und berufsethischen Fragen. Die EKs legen ihrer Arbeit die gesetzlichen Bestimmungen und berufsrechtlichen Rahmenbedingungen sowie die Deklaration von Helsinki des Weltärztebundes zugrunde. Einer EK muss jedes Forschungsvorhaben an Menschen oder an menschlichem Blut oder Gewebe vorgelegt werden. Die Hauptaufgabe der EK besteht also in der Wahrnehmung einer ethischen und rechtlichen Verantwortung für Patienten und Probanden. Hierbei wägt die EK ab zwischen zwei verfassungsgemäß garantierten Grundrechten, einerseits dem Grundrecht jedes Bürgers auf Unversehrtheit und andererseits der Freiheit von Wissenschaft und Forschung. Hierbei hat sie dem Wohl des einzelnen Patienten oder Probanden stets Vorrang einzuräumen. Da die klinische Forschung dem medizinischen Fortschritt dient, muss die Ethikkommission also abwägen zwischen dem Wohl des Studienteilnehmers und dem Wohl von Patienten, die in Zukunft mit der zu erprobenden Therapie erfolgreich behandelt werden könnten. Diese diffizile Aufgabe einer EK wird durch die Abbildung 1 verdeutlicht. |
Es gibt keine scharfe Abgrenzung der einzelnen Phasen voneinander in den Gesetzestexten. Zwar existieren einzelne absolute Kriterien, jedoch auch viele fließende Übergänge. Absolute Kriterien sind:
Phase I: gesunde Probanden (keine Patienten) zum Test der Verträglichkeit;
Phase II: Bestätigung des Therapiekonzeptes (proof of concept) im Hinblick auf die Wirksamkeit;
Phase III: pivotale (entscheidende) Zulassungsstudie(n);
Phase IV: nach der Zulassung.
Voraussetzungen für klinische Studien
Die Voraussetzungen, die erfüllt sein müssen, um mit einer klinischen Studie beginnen zu können, sind im AMG geregelt. Im Einzelnen:
Es muss einen Sponsor (trägt die Verantwortung für die Veranlassung, Organisation und Finanzierung) mit Sitz in der EU geben (notfalls kann ein gesetzlicher Vertreter benannt sein).
Die Verantwortung für die klinische Prüfung muss bei einem verantwortlichen Prüfer liegen, der durch den Sponsor benannt wird.
Für die Studienteilnehmer (Patienten und Probanden) muss eine Versicherung abgeschlossen werden.
Die Einrichtung, in der die klinische Prüfung stattfinden soll, muss geeignet sein.
Die Prüfer müssen adäquat qualifiziert sein.
Eine vorangegangene präklinische Testung am Tier muss vorliegen (siehe 1. Teil dieser Serie).
Ein Arzt muss die medizinische Versorgung der Studienteilnehmer sicherstellen.
Weitere Voraussetzungen sind
ein zustimmendes Votum durch die Ethikkommission (siehe Infobox und Abb. 1),
die Genehmigung durch die zuständige Bundesoberbehörde,
eine Anzeige bei den zuständigen Landesbehörden,
eine positive Nutzen-Risiko-Abwägung.
Infobox: Deklaration von Helsinki und ethisches HandelnDie Deklaration von Helsinki (Declaration of Helsinki, DoH) ist ein medizinischer Verhaltenskodex; sie enthält ethische Leitlinien für Ärzte im Umgang mit Patienten und Probanden. Unter dem Eindruck der grausamen Menschenversuche während des 2. Weltkrieges wurde sie vom Weltärztebund (World Medical Association, WMA) 1960 in einer ersten Form verfasst und bis heute immer wieder aktualisiert und den neuen Gegebenheiten der klinischen Forschung angepasst. Die DoH ist der international anerkannte Stand der Ethik in der Medizin und gilt als unabdingbare Grundlage einer lege artis geplanten klinischen Studie, insbesondere bei der vorangehenden Beurteilung durch die Ethikkommission (soft law). Die DoH folgt im Wesentlichen zwei entscheidenden Grundsätzen. Zum einen beruht Fortschritt in der Medizin auf Forschung. Klinische Forschung, d. h. Forschung am Menschen sollte daher unterstützt werden, soweit sie ethisch vertretbar ist. Der zweite Grundsatz besagt, dass das Wohlergehen der Versuchsperson Vorrang hat vor den Interessen von Wissenschaft, Gesellschaft und Wirtschaft. Dabei soll die Verantwortung für die Versuchsperson stets in den Händen eines Arztes liegen. Wichtig sind die weitreichenden Rechte der Versuchsperson: Privatsphäre und Datenschutz sind strengen Regeln unterworfen, die eingehalten werden müssen. Es darf einem Außenstehenden nicht möglich sein, eine Versuchsperson zu identifizieren. Besondere Schutzrechte gelten beispielsweise für Minderjährige oder kognitiv eingeschränkte Personen. Zwingende Voraussetzung für die Teilnahme an einer klinischen Studie ist die umfassende Aufklärung des Prüfungsteilnehmers (informed consent). Dieser kann das nach der Aufklärung schriftlich gegebene Einverständnis jederzeit ohne Angabe von Gründen widerrufen. |
Das Genehmigungsverfahren
Ein Antrag auf Genehmigung einer klinischen Prüfung muss nicht nur der federführenden Ethikkommission vorgelegt werden, sondern auch bei der zuständigen Bundesoberbehörde durch den Sponsor eingereicht werden. Die rechtliche Grundlage zu diesem Genehmigungsverfahren ist im § 40 des AMG sowie im § 7 der GCP-Verordnung festgelegt. Hier sind auch die Angaben aufgelistet, die der Sponsor bei der Beantragung einer klinischen Studie machen muss.
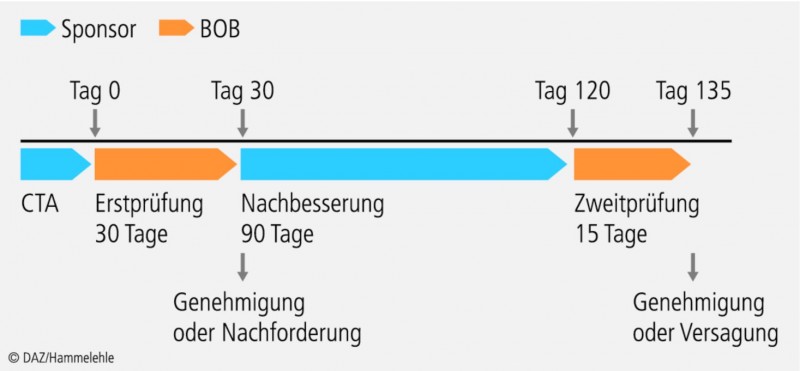
Vorher muss die klinische Studie in der EudraCT-Datenbank (eine Datenbank in der alle klinischen Studien in der EU erfasst werden) registriert werden, und zum Beleg hierfür ist der Bundesoberbehörde die EudraCT-Nummer mitzuteilen. Einen Überblick über die zeitliche Abfolge des Genehmigungsverfahrens gibt Abbildung 2.
Es gilt das Prinzip der impliziten Genehmigung, das heißt: Wenn die zuständige Bundesoberbehörde nicht im vorgegebenen Zeitfenster reagiert, indem sie eine Nachforderung stellt oder die Studie untersagt, gilt die Genehmigung als erteilt.
Der Prüfplan
Das Herzstück eines Antrags zur Genehmigung einer klinischen Prüfung von Arzneimitteln ist der Prüfplan, weil er die Grundlage der Entscheidung bildet, ob die Genehmigung erteilt wird. Der Prüfplan wird auch als Studienprotokoll (engl. study protocol) bezeichnet. Seine rechtliche Grundlage ist das AMG und die Deklaration von Helsinki (siehe Infobox oben), die beide vor der Rekrutierung von Patienten die Vorlage eines Prüfplans fordern.
Der Prüfplan muss sowohl der zuständigen Bundesoberbehörde als auch der Ethikkommission vorgelegt werden. Sein Inhalt ist beschrieben in der GCP-Leitlinie (siehe Infobox). Ein Prüfplan muss nach GCP (mindestens) die folgenden Informationen enthalten:
Studiensynopse,
Nutzen-Risiko-Bewertung,
Einschlusskriterien,
Ausschlusskriterien,
- Abbruchkriterien,
Bewertungskriterien der Wirksamkeit,
Bewertungskriterien der Sicherheit,
Angaben zu Biometrie und Statistik,
Angaben darüber, wo und wie die zuständige Bundesoberbehörde Zugang zu den Originaldaten und Originaldokumenten bekommt,
Maßnahmen zur Qualitätssicherung,
eine ethische Einschätzung des Vorhabens,
Datenmanagement,
Angaben, wie der Sponsor seiner Aufbewahrungspflicht der Studiendokumentation nach AMG nachkommen will,
Angaben, wie die Finanzierung der Studie zustande kommt und
Angaben über den Abschluss der Probanden-/Patientenversicherung (über mind. 500.000 Euro im extremen Schadensfall).
Studiendesign
Bevor auf die Phasen der klinischen Prüfung eingegangen wird, sollen hier kurz die wichtigsten Begriffe, mit denen klinische Studien gekennzeichnet werden, am Beispiel der folgenden (fiktiven) Studie erklärt werden:
"An open label, balanced, randomized, single-dose, cross-over, placebo-controlled study of substance X in healthy, adult, human subjects under fasting conditions."
In diesem Fall soll ein neuer Wirkstoff X (Prüfsubstanz, Verum) im Vergleich mit Placebo an gesunden Erwachsenen (Probanden, keine Patienten) unter folgenden Bedingungen getestet werden:
open label bedeutet, dass sowohl der Proband als auch der Arzt wissen, welcher Proband welche Medikation erhält (Placebo oder Verum).
balanced bedeutet, dass beide Gruppen (Studienarme) gleich groß sind. Während einer klinischen Studie kommt es immer wieder zu dropouts (Probanden/Patienten, die die Studie verlassen) oder withdrawls (Probanden/Patienten, die der Leiter der klinischen Prüfung beispielsweise aus Sicherheitsgründen aus der Studie zurückzieht). Dies führt dazu, dass am Ende einer Studie fast immer weniger Teilnehmer vorhanden sind, als ursprünglich eingeschlossen wurden. Der Begriff "balanced” bezieht sich also auf das geplante, nicht auf das tatsächliche Zahlenverhältnis der Gruppen.
randomized bedeutet, dass die Probanden zufällig auf die beiden Gruppen verteilt werden. Die Randomisierung wird heute oft per Computer durchgeführt. Sie beugt einem systematischen Fehler vor, der beispielsweise entstehen würde, wenn ältere Patienten nur der einen Gruppe zugeordnet werden würden. Das würde nämlich bedeuten, dass man mit einer Studie zwei Variablen untersucht, z. B. alte Patienten & Placebo vs. junge Patienten & Verum.
single-dose bedeutet, dass die Probanden die Prüfsubstanz nur einmal erhalten.
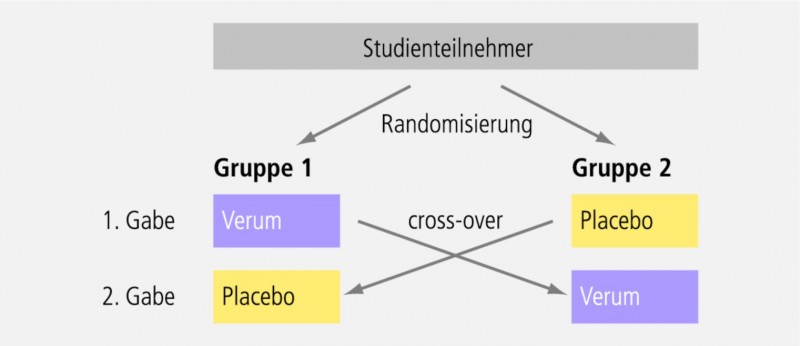
cross-over bedeutet, dass beide Studienarme beide Medikationen bekommen, allerdings in unterschiedlicher Reihenfolge. Zweck eines Cross-over-Designs ist es, eventuelle Unterschiede zwischen den beiden Studienarmen bestmöglich zu kompensieren (intraindividuelle Variabilität; s. Abb. 3).
Meistens soll die klinische Prüfung eines neuen Arzneistoffs beweisen, dass Sicherheit und Wirksamkeit besser (überlegen) sind als die bisher auf dem Markt befindlichen Arzneistoffe mit gleicher Indikation. Andere Studien sollen lediglich beweisen, dass der fragliche Arzneistoff in etwa gleich gut (äquivalent) oder nicht schlechter (nicht unterlegen) ist als eine Vergleichsmedikation.
Die Definitionen nach ICH stehen im Textkasten.
Definitionen nach ICHÜberlegenheitsstudie (Superiority Trial) A trial with the primary objective of showing that the response to the investigational product is superior to a comparative agent. Äquivalenzstudie (Equivalence Trial) A trial with the primary objective of showing that the response to two or more treatments differs by an amount which is clinically unimportant. Nicht-Unterlegenheitsstudie (Non-Inferiority Trial) A trial with the primary objective of showing that the response to the investigational product is not clinically inferior to a comparative agent. Quelle: ICH E9-Guideline, Glossary: http://firstclinical.com/regdocs/doc/?showpage=62&db=ICH_E9 |
Endpunkte in klinischen Studien
Der Endpunkt einer klinischen Studie beschreibt ihr Zielkriterium. Die Endpunkte von prospektiv geplanten klinischen Studien müssen vor Beginn der Rekrutierung im Studienplan (und ‑protokoll) festgelegt werden. Einen klinischen Endpunkt definiert die ICH wie folgt: "clinical endpoint: A characteristic or variable that reflects how a patient feels, functions or survives."
Primäre (harte) Endpunkte sind z. B. Heilung, Überleben oder Morbidität.
Sekundäre Endpunkte sind z. B. Symptome und Laborparameter, die zur Beurteilung des Erfolgs ebenfalls von Bedeutung sind.
Zudem können Surrogatendpunkte festgelegt werden. Ein Surrogatendpunkt ist eine Variable, die relativ einfach zu messen ist und die das Ergebnis einer therapeutischen Intervention vorhersagt, ohne selbst ein direkter Maßstab für den klinischen Nutzen oder Schaden zu sein.
Die Identifikation der passenden Endpunkte gehört zu den schwierigsten Aufgaben bei der Planung einer klinischen Studie. Zur Beratung in diesem Punkt können Gespräche mit der zuständigen Bundesoberbehörde und (hinsichtlich der frühen Nutzenbewertung) mit dem G‑BA und dem IQWiG beantragt werden.
Phase 0
Klinische Studien der Phase 0 sind im Gegensatz zu den Phasen I bis III ein gesetzlich nicht vorgeschriebenes Verfahren. Es handelt sich dabei um eine Testung der pharmakokinetischen Eigenschaften eines neuen Wirkstoffes per Microdosing. Das heißt, dass der Wirkstoff weit unterhalb der Schwellendosis für eine pharmakodynamische Wirkung verabreicht wird. Dies geschieht in der Regel an gesunden Probanden. Moderne, hochempfindliche Analysenverfahren geben hierbei Erkenntnisse zur Wirkstoffverteilung und Metabolisierung. Als Analytik kommt beispielsweise die Massenspektroskopie infrage nach chromatografischer Auftrennung. Zusammengenommen besteht die Phase 0 also aus einer Voruntersuchung zur optimalen Vorbereitung der pharmakokinetischen Daten und der Sicherheitsdaten der Phase I.
Phase I
Getestet wird in der Regel an gesunden, männlichen, jungen Probanden (ab 18 Jahre). Ziele sind Verträglichkeit, Pharmakokinetik, Pharmakodynamik und die Dosisfindung für die Patienten in Phase II. Hierzu wird die Phase I oft als Dosiseskalationsstudie durchgeführt.
Eine Dosiseskalationsstudie wird üblicherweise nach folgendem Verfahren durchgeführt. Den Einstieg in die klinische Studie der Phase I bildet die Maximum Recommended Starting Dose (MRSD), also eine aus der präklinischen Toxikologie abgeleitete Initialdosis (siehe hierzu den 1. Teil dieser Serie). Die MRSD wird einer Gruppe von drei Probanden verabreicht. Anschließend wird die doppelte Dosis einer weiteren Gruppe von drei Patienten verabreicht. Diese Prozedur der Dosisverdopplung vollzieht sich in der Regel dreimal, sodass insgesamt vier Gruppen von jeweils drei Probanden untersucht werden.
Die Phase I erstreckt sich meist über einen Zeitraum von wenigen Tagen. Wenn möglich und ethisch vertretbar, wird sie als Cross-over-Design durchgeführt (Abb. 3), wobei die beiden Behandlungsperioden von einer Wash-out-Phase getrennt sind. Die Wash-out-Phase soll einen vollständigen metabolischen Abbau des Testmedikamentes aus der ersten Gabe sicherstellen und mindestens fünf Plasmahalbwertszeiten (oft deutlich mehr) betragen. Die Probanden der Phase I sind hospitalisiert. Sie werden sehr exakt und in häufigen Intervallen überwacht, nicht nur hinsichtlich der Blutentnahme, sondern auch was die Vitalzeichen angeht: Blutdruck, Herzfrequenz, EKG etc.; zudem ist bei Bedarf eine notfallmedizinische Intensivversorgung sichergestellt.
Ein häufiges Missverständnis sind die Ziele der Phase I der klinischen Prüfung: Es geht hier nicht um die Wirksamkeit der Prüfsubstanz, denn die Probanden sind ja nicht krank. Es können höchsten Surrogatparameter der Wirksamkeit (Blutdruck, Pulsfrequenz, Blutbild, klinische Chemie etc.) erfasst werden.
Besondere Anforderungen an Phase-I-Studien
Bei Untersuchungen von innovativen Arzneimitteln in der Phase I wird naturgemäß großes Augenmerk auf das Risikopotenzial der Prüfsubstanz gelegt. In diesem Zusammenhang soll der Sponsor der Studie sich zu der Frage äußern, ob es sich um eine sogenannte Hochrisikosubstanz handelt. Es muss mindestens eine der folgenden Bedingungen vorliegen, damit es sich um eine Hochrisikosubstanz handelt:
Der Wirkmechanismus ist nicht genau bekannt.
Die Funktion der Zielstruktur ist nicht genau bekannt.
Die Relevanz der Tierversuche in der Präklinik ist schwer abschätzbar.
Für Wirkstoffe, die unter die Hochrisikokriterien fallen, sollte die Dosis sich nicht mehr wie bisher ausschließlich am "No Observed Adverse Effect Level" (NOAEL), sondern am "Minimal Anticipated Biological Effect Level" (MABEL) orientieren (siehe 1. Teil dieser Serie). Die Startdosis, die Dosisstufen und die maximale Dosis müssen detailliert begründet werden.
Beim ersten Einsatz am Menschen sollte geprüft werden, ob eine intravenöse Gabe notwendig ist – wenn ja, dann sollte der Wirkstoff langsam appliziert werden, um die Infusion im Bedarfsfall rasch unterbrechen zu können, falls schwerwiegende Nebenwirkungen auftreten.
Zudem sollten nicht mehrere Probanden gleichzeitig exponiert werden (sequenzielles Design). Weiterhin sollte im Prüfplan detailliert beschrieben werden, unter welchen Voraussetzungen eine Dosissteigerung erfolgt und welche Maßnahmen im Falle eines Notfalls durchgeführt werden (z. B. Glucocorticoid-Gabe, Dialyse o. ä.).
Kombinierte Phase I/II
Ist die Ersterprobung eines neuen Arzneistoffes an gesunden Probanden ethisch nicht vertretbar, so werden bereits die frühen zulassungsrelevanten Studien an Patienten durchgeführt. Dies bezieht sich vor allem auf Zytostatika (Chemotherapeutika), da es in diesem Bereich zu massiven Nebenwirkungen kommt und kanzerogene Spätfolgen auftreten können. Auch andere Wirkstoffe mit schweren Nebenwirkungen werden nicht an gesunden Probanden erprobt.
Phase II
Klinische Prüfungen der Phase II führen den ersten Nachweis der medizinischen Wirksamkeit eines innovativen Medikamentes (efficacy explorativ). Phase-II-Studien werden üblicherweise an 200 bis 400 Patienten mit der avisierten Indikation des Medikamentes durchgeführt. Hier erfolgt also der erste Einsatz des Medikamentes in der Zielpopulation. Die Ein- und Ausschlusskriterien werden in Phase II streng gehalten, sodass homogene Patientenkollektive entstehen. Die Behandlungsdauer ist in der Regel auf wenige Monate beschränkt. Ziel ist die Bestätigung des Therapiekonzeptes, das sogenannte proof of concept (POC).
Parallel mit der medizinischen Wirksamkeit erfolgt eine weitere Prüfung der Verträglichkeit. Falls die initiale Phase-II-Studie eine gute Wirksamkeit ergibt, erfolgt eine weitere Teilstudie, die sogenannte Phase IIb. Hierbei geht es darum, die optimale therapeutische Dosis für die anschließenden pivotalen Phase-III-Studien herauszufinden. In Phase II werden zudem oftmals Surrogatendpunkte verwendet und nicht (harte) klinische Endpunkte. Gegebenenfalls erfolgt während der Phase II auch eine Optimierung bzw. ein Wechsel der Arzneiform. Ist das der Fall, so müssen nochmals Phase-I-Studien zur Pharmakokinetik und -dynamik mit der neuen Formulierung durchgeführt werden.
Phase III
Für die Erstellung eines Zulassungsdossiers ist die Phase III der wichtigste (pivotale) Teil der klinischen Prüfung. Ziel ist der endgültige Beweis der Wirksamkeit (efficacy konfirmatorisch). Aber auch der Nachweis eines therapeutischen Zusatznutzens im Vergleich zur zweckmäßigen Vergleichstherapie nach AMNOG rückt nun vermehrt in den Fokus. Die Grundlage für die Zulassung entsteht, indem die Wirksamkeit und Unbedenklichkeit, die die Prüfsubstanz in der positiv verlaufenen Phase II gezeigt hat, an großen Patientenkollektiven (mehrere Hundert bis mehrere Tausend) bestätigt werden. Die Phase III kann sich über mehrere Monate bis hin zu vielen Jahren erstrecken. Sie wird in der Regel in einem randomisierten, kontrollierten, verblindeten (doppel- oder dreifachblinden) Studiendesign durchgeführt.
Oft wird auch die Pharmakokinetik bei Patienten mit Nieren- oder Leberinsuffizienz, bei Senioren, Kindern und anderen besonderen Patientengruppen (special populations) untersucht, um herauszufinden, ob eine Dosisanpassung bei diesen Gruppen notwendig ist.
Phase IV
Unter dem allgemeinen Begriff der Phase IV wird jede Art der klinischen Arzneimittelforschung zusammengefasst, die nach der Zulassung ansetzt (daher der englische Begriff post marketing authorisation studies).
Die Fragestellungen an Phase-IV-Untersuchungen und die daraus resultierenden Formen der Studien sind vielfältig:
Anwendungsbeobachtungen zur Untersuchung von seltenen und sehr seltenen Nebenwirkungen. Sie lassen sich nur bei sehr großen Fallzahlen statistisch signifikant identifizieren.
Anwendungsbeobachtungen zur Untersuchung von Interaktionen, insbesondere bei älteren und/oder multimorbiden Patienten mit u. U. vielfältiger Begleitmedikation.
Patientenrelevanter Nutzen.
Pharmakoökonomie: frühe Nutzenbewertung, Nutzenbewertung, Kosten-Nutzenbewertung (HTA, Health Technology Assessment).
Produktlinienerweiterung.
Ein Sonderfall sind Indikationserweiterungen. Hierbei handelt es sich zwar um Untersuchungen an einem bereits zugelassenen Medikament, jedoch liegt keine Zulassung für die angestrebte Indikation vor, sodass es sich streng genommen wiederum um eine Phase-III-Zulassungsstudie handelt.
Eine große Gruppe bilden nach der Zulassung die wissenschaftsinitiierten Studien (IITs, investigator initiated trials).
Anwendungsbeobachtungen
Eine besondere Form der Phase-IV-Studien ist die Anwendungsbeobachtung. Hierbei handelt es sich nicht um eine klinische Prüfung nach AMG, denn es erfolgen keine über die normale Therapie hinausgehenden Eingriffe und Untersuchungen oder Interventionen am Patienten. Es handelt sich also um eine nicht-interventionelle Studie (NIS). Bei der Anwendungsbeobachtung geht es um eine genaue Beobachtung im routinemäßigen Einsatz des Medikamentes. Diese Beobachtung erfolgt nach epidemiologischen Methoden ohne einen vorher festgelegten Prüfplan – darin besteht der wichtigste Unterschied zu den Phasen I bis III. Die Anwendungsbeobachtung kann der Pharmakovigilanz oder der besseren Vermarktung des Medikaments dienen.
Infobox: Die Good Clinical Practice (GCP)-Leitlinie der ICHDie GCP-Leitlinie der ICH umfasst Regeln zur Durchführung klinischer Prüfungen nach international anerkannten Standards und dem Stand von Wissenschaft und Technik. Sie dient der Sicherheit und dem Schutz der Prüfungsteilnehmer (Probanden bzw. Patienten) und sichert die Qualität der erhobenen Daten. Ihren Ursprung hat die GCP-Leitlinie in der Erkenntnis, dass kontrollierte Zulassungsverfahren in den Industrienationen harmonisiert werden sollen. Lange galten unterschiedliche Richtlinien in den USA, Europa und Japan. Da sich die Pharmaindustrie bereits der globalen Herausforderung gestellt hatte, wurde eine Rationalisierung und Harmonisierung nötig: 1990 wurde die ICH gegründet (International Conference on Harmonisation of Technical Requirement for the Registration of Pharmaceuticals for Human Use). Das Ergebnis war 1997 die Verabschiedung der ICH E6 (R1) Guideline for Good Clinical Practise, kurz als GCP-Guideline oder GCP-Leitlinie bezeichnet. Von der European Medicines Agency (EMA) in das System europäischer Leitlinien aufgenommen, heißt sie in Europa auch: CPMP/ICH/135/95. Die GCP-Leitlinie gilt u. a. in den USA, in Kanada, EU-Europa und Japan, das heißt, dass die Zulassungsbehörden in diesen Staaten klinische Prüfungen nur anerkennen, wenn sie GCP-konform durchgeführt worden sind. Die GCP-Leitlinie sieht vor, dass der Prüfarzt die Deklaration von Helsinki des Weltärztebundes in ihrer jeweils aktuellen Fassung einzuhalten hat. Hierin haben sich die Ärzte zu ethischen Grundsätzen bei medizinischem Handeln verpflichtet. Zudem müssen sich alle Beteiligten den Gesetzen des Landes unterordnen, in dem die klinische Studie durchgeführt wird. Die GCP-Leitlinie schreibt eine Nutzen-Risiko-Abwägung für jeden einzelnen Prüfungsteilnehmer vor (Einzelfallbetrachtung). Die Rechte, die Sicherheit und das Wohl der Prüfungsteilnehmer haben Vorrang vor den Interessen von Wissenschaft und Gesellschaft. 1997 war die GCP-Guideline zunächst "nur" eine Leitlinie zur gegenseitigen Anerkennung von Studienergebnissen der ICH-Länder. 2001 regelte eine Richtlinie der EU (2001/20/EG) die Implementierung der GCP-Guideline in die nationale Gesetzgebung. Die Umsetzung in Deutschland erfolgte 2004 mit der 12. AMG-Novelle (GCP-Verordnung). |
Dokumentation und Management von Nebenwirkungen
Das Nebenwirkungsmanagement bei klinischen Prüfungen gehört naturgemäß zu den komplexesten Problemen, die Prüfer und Sponsor zu bewältigen haben. Grundsätzlich sind zwei Arten von unerwünschten Ereignissen zu unterscheiden:
Adverse events (AE), alle Ereignisse, die einem Probanden oder Patienten während der klinischen Prüfung widerfahren, unabhängig davon, ob sie in Zusammenhang mit dem Prüfpräparat stehen oder nicht.
Adverse reactions (AR), Ereignisse, die einem Probanden oder Patienten während der klinischen Prüfung widerfahren und in einem Zusammenhang mit dem Prüfpräparat stehen (Abb. 4).
Dokumentiert werden alle AEs. Anschließend äußert der Prüfarzt seine Einschätzung, ob diese in einem Zusammenhang mit der Prüfmedikation stehen. Abgesehen von der Kausalität bewertet der Prüfer auch den Schweregrad einer AE oder AR. Mit "schwerwiegend" (engl. serious) sind nach der EU-Direktive 2001/20/EC Ereignisse gemeint, die
• zum Tod führen,
• lebensbedrohlich sind,
• eine Hospitalisierung des Studienteilnehmers erforderlich machen,
• zu einer dauerhaften Behinderung oder Invalidität führen und
• zu Geburtsfehlern führen.
Im nächsten Schritt wird die "expectedness" einer AE oder AR bewertet, d. h. ob ein unerwünschtes Ereignis aufgrund der bereits vorliegenden präklinischen oder klinischen Daten als "erwartet" oder "unerwartet" eingestuft wird. Bei bereits zugelassenen Arzneimitteln werden Unexpected SAR (USAR), also unerwartete schwerwiegende unerwünschte Wirkungen, definiert als Nebenwirkungen, deren Art, Ausmaß oder Ausgang von der Packungsbeilage des Arzneimittels abweichen bzw. nach GCP-Verordnung nicht mit der vorliegenden Information über das Prüfpräparat übereinstimmen (§ 4 Abs. 13 AMG). Bei einer Prüfsubstanz ist aber zunächst fraglich, ob sie diese unerwartete Reaktion hervorgerufen hat. Daher wurde der Begriff "Verdachtsfall einer unerwarteten schwerwiegenden Nebenwirkung” eingeführt (engl. suspected unexpected serious adverse reaction, SUSAR). Diese Verdachtsfälle bedürfen einer sofortigen eingehenden Betrachtung durch Prüfarzt und Sponsor. Sie müssen innerhalb von 15 Tagen an die zuständige Behörde und die federführende Ethikkommission gemeldet werden. Wenn ein Patient gestorben ist, beträgt die Meldefrist sieben Tage.
Quellen
Deutsches Arzneimittelgesetz (AMG); www.gesetze-im-internet.de/amg_1976.
GCP-Verordnung; www.gesetze-im-internet.de/gcp-v/index.html.
Regulatorischen Texte zu klinischen Prüfungen in der EU. Eudralex 10; http://ec.europa.eu/health/documents/eudralex/vol-10.
Geplante Studien: www.clinicaltrials.org oder https://eudract.ema.europa.eu.
Heinzl S. Klinische Studien: Wie Arzneimittel geprüft werden. Pharm Ztg 2011;156(30):16 – 20.
Autoren
Dr. Niels Eckstein (korrespondierender Autor), Mirabellenstr. 12, 53340 Meckenheim, Niels.Eckstein@web.de
Dr. Bodo Haas, bodohaas@web.de
Weitere Informationen zu den Autoren in DAZ Nr. 25, S. 76



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.