












- DAZ.online
- DAZ / AZ
- DAZ 43/2012
- Neue Sicherheitsmerkmale ...
Arzneimittelssicherheit
Neue Sicherheitsmerkmale gegen Fälschungen
Foto: IFA
Als Bestandteil des Pharma Package ist seit dem 21. Juli 2011 die unter dem Schlagwort "Fälschungsrichtlinie" bekannte Richtlinie 2011/62/EU des Europäischen Parlaments und des Rates vom 8. Juni 2011 in Kraft. In Deutschland sind deren Inhalte jüngst durch die sogenannte 16. AMG-Novelle umgesetzt worden, deren Verkündung kurz bevorsteht.
Kernstück der Novelle: neue Sicherheitsmerkmale
Die Novellierungen zum Schutz der Verbraucher vor Arzneimittelfälschungen sind zahlreich und greifen an verschiedenen Stellen der Vertriebskette. Das Kernstück der Novelle bildet die Einführung von neuen Sicherheitsmerkmalen, die Thema dieses Beitrages sind. Insoweit wird in Zukunft der Aufdruck eines Identifikations- und eines Anti-Manipulationsmerkmales obligatorisch.
Definition von "Arzneimittelfälschung"
Vor dem Erlass der Fälschungsrichtlinie wurden zunächst nationale Unterschiede in Bezug auf die Definition der Arzneimittelfälschung festgestellt. Um diesbezüglich Klarheit zu schaffen, führte man eine Definition entsprechend § 4 Absatz 40 Arzneimittelgesetz (AMG) ein; von einem gefälschten Arzneimittel ist danach auszugehen, wenn es falsche Angaben über seine Identität, Herkunft oder den Vertriebsweg enthält. Damit fallen nunmehr EU-weit weder versehentliche Qualitätsabweichungen noch die Verletzung geistiger Schutzrechte wie Marken- oder Patentverletzungen unter die Definition des gefälschten Arzneimittels.
Ausgestaltung der Sicherheitsmerkmale
Die Fälschungsrichtlinie sieht bei Arzneimitteln zum einen ein individuelles Erkennungsmerkmal (unique identifier), zum anderen ein Anti-Manipulationsmerkmal (tamper proof evidence) vor. Idee des Erkennungsmerkmals ist es, der gesamten Vertriebskette – nämlich dem Großhandel, der Apotheke und weiteren beteiligten Personen – die Möglichkeit zu geben, die Echtheit des Arzneimittels zu verifizieren und jede einzelne Arzneimittelpackung bei der Abgabe zu identifizieren. Das Anti-Manipulationsmerkmal soll gewährleisten, dass aufgrund einer Sichtprüfung festgestellt werden kann, ob ein Arzneimittel gefälscht ist bzw. ob an einem originalen Arzneimittel Manipulationen vorgenommen oder versucht wurden.
Der im Rahmen der Novelle neu gestaltete § 10 Absatz 1c AMG bestimmt diesbezüglich, dass "bei Arzneimitteln, die zur Anwendung bei Menschen bestimmt sind, (...) auf den äußeren Umhüllungen Sicherheitsmerkmale sowie eine Vorrichtung zum Erkennen einer möglichen Manipulation der äußeren Umhüllung anzubringen" sind.
Welcher Form und welchem Inhalt diese Sicherheitsmerkmale entsprechen müssen, regelt das Gesetz nicht. Der europäische Richtliniengeber hatte früh erkannt, dass bei der Umsetzung derartiger Systeme zur Bekämpfung von Arzneimittelfälschungen die Kenntnisse und Erfahrungen der betroffenen Verkehrskreise unerlässlich sind. Aufgrund dessen wurden die Details der Regelungen nicht in die Richtlinie aufgenommen, sondern vielmehr der Europäischen Kommission zur Ausarbeitung übertragen. Diese soll im Rahmen von sogenannten delegierten Rechtsakten – vergleichbar den deutschen Rechtsverordnungen, z. B. der Apothekenbetriebsordnung – einen umsetzbaren Rechtsrahmen schaffen und die von der Richtlinie vorgezeichneten Maßnahmen zur Einführung von Sicherheitsmerkmalen umsetzen. Eine Veröffentlichung der Rechtsakte wird für das Jahr 2014 erwartet; bislang hat die Kommission keine Inhalte festgelegt.
Sobald die Modalitäten in den delegierten Rechtsakten festgeschrieben sind, sind diese – nach einem Übergangszeitraum – in das nationale Recht zu übernehmen.
black list & white list
Die Sicherheitsmerkmale sollen nach dem Willen der Richtlinie nicht ausnahmslos für alle Arzneimittel gelten: Grundsätzlich sind nur verschreibungspflichtige Arzneimittel vom Anwendungsbereich erfasst. Ausnahmen gelten dabei in beide Richtungen: Es können nicht verschreibungspflichtige Arzneimittel der Pflicht zum Tragen von Sicherheitsmerkmalen unterstellt werden (sog. black list), und ebenso können verschreibungspflichtige Arzneimittel von dieser Pflicht befreit werden (white list).
Welche Arzneimittel in die entsprechenden Listen eingeordnet werden, ist wiederum Regelungsgegenstand der delegierten Rechtsakte. Die Kriterien für die Einordnung gibt die Richtlinie 2001/83/EG in Artikel 54a Absatz 2b vor. Es sollen berücksichtigt werden:
Preis und Absatzvolumen des Arzneimittels,
Anzahl und Häufigkeit der in der Vergangenheit gemeldeten Fälle von gefälschten Arzneimitteln in der Union und in Drittländern,
Schweregrad der zu behandelnden Erkrankung und
das Risiko aufgrund geringen Fälschungsaufwands.
Der Umfang der Liste ist derzeit nicht absehbar. Denkt man an frühere nationale Bestrebungen der Einordnung von Arzneimitteln in Positiv- bzw. Negativ-Listen und an die Vorgaben des Gemeinsamen Bundesausschusses betreffend die Erstattungsfähigkeit von Arzneimitteln und Medizinprodukten nach § 31 Absatz 1 Satz 2 und 3 SGB V, dürfte das Verfahren zu ausgiebigen Diskussionen Anlass geben. Gerade im Fall der "black list" dürfte es aufgrund der ausnahmsweisen Kostenbelastung der pharmazeutischen Unternehmer zu Streitigkeiten kommen.
Pilotprojekt securPharm
Um der Europäischen Kommission ein funktionierendes und sinnvolles Pilotmodell zur Umsetzung der Sicherheitsvorgaben bzgl. der Arzneimittel-identifizierung zu demonstrieren und den delegierten Rechtsakten gewissermaßen zuvorzukommen, haben die maßgeblichen Verbände der Pharmaindustrie, des Großhandels und der Apotheker den Verein "securPharm" gegründet. Ziel des Modells ist es, ein möglichst sicheres, aber auch praktikables und kosteneffizientes Modell zur Umsetzung der neuen Sicherheitsvorgaben zu schaffen. Mitglieder sind die Bundesvereinigung Deutscher Apothekerverbände (ABDA), der Bundesverband der Arzneimittelhersteller e. V. (BAH), der Bundesverband der Pharmazeutischen Industrie e. V. (BPI), der Bundesverband des pharmazeutischen Großhandels (Phagro), Pro Generika e. V. und der Verband Forschender Arzneimittelhersteller e. V. (vfa).
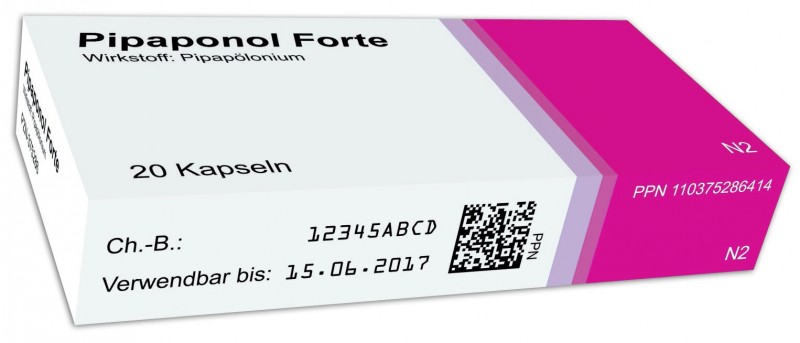
Das Pilotprojekt, das Anfang des Jahres 2013 starten soll und für zunächst drei Monate geplant ist, hat sich (nur) der Ausgestaltung des Identifizierungsmerkmals verschrieben. Es sieht die Einführung eines DataMatrix-Codes, den Betrieb einer Datenbank durch die Stakeholder und eine End-to-end-Verifizierung durch die abgebende Apotheke vor.
DataMatrix-Code
Die bisherige Produktkennzeichnung, bestehend aus PZN (Klarschrift und EAN-Strichcode) sowie Charge und Verfallsdatum (Klarschrift), soll künftig durch den DataMatrix-Code ersetzt werden. Geplant ist die Angabe der Pharmacy-Product-Number (PPN, Klarschrift und codiert), der Charge und des Verfallsdatums (Klarschrift und codiert) sowie einer randomisierten Seriennummer (codiert). Die Seriennummer wird vom pharmazeutischen Unternehmer vergeben, ist einmalig und darf nicht deterministisch (vorher festgelegt) sein.
Datenbanksystem für End-to-end-Verifizierung
Weiter sieht securPharm die Einführung eines neuen Datenbanksystems vor, das den Zyklus eines Arzneimittels bis zur Abgabe an den Patienten verfolgen soll (End-to-end-Verifizierung).
Der pharmazeutische Unternehmer trägt jedes mit einer individuellen Seriennummer versehene Arzneimittel in eine geschützte Datenbank (Herstellerdatenbank) ein und markiert es im Falle des Verkaufs (z. B. an den Großhandel) als in den Markt gegeben. Soll ein Arzneimittel nun in der Apotheke abgegeben werden, gleicht das Datenbanksystem der Apotheken (ABDATA) die Angaben des eingescannten Produktes mit der Herstellerdatenbank ab.
Wenn die Datenbank eine Kongruenz feststellt und das Arzneimittel in der Herstellerdatenbank als noch nicht abgegeben vermerkt ist, kann das Produkt dem Verbraucher übergeben werden; es wird dann als abgegeben gekennzeichnet.
Wenn das ABDATA-System kein unter dem eingescannten DataMatrix-Code eingetragenes Produkt findet oder dieses als bereits abgegeben gekennzeichnet ist, verbietet sich eine Abgabe. Vielmehr wird der Apotheker mit einem Warnhinweis auf eine mögliche Fälschung hingewiesen.
Zeitlicher Rahmen der Änderungen
Die Umsetzung des neuen Systems in die Praxis einschließlich der Schaffung einer entsprechenden Datenbank wird einige Zeit in Anspruch nehmen; dem hat auch der Richtliniengeber Rechnung getragen, denn § 10 Absatz 1c AMG, der die Sicherheitsmerkmale auf der äußeren Umhüllung verlangt, tritt erst drei Jahre nach Veröffentlichung der delegierten Rechtsakte in Kraft. Nach dem aktuell vorhersehbaren Ablauf werden die delegierten Rechtsakte wahrscheinlich im Jahr 2014 veröffentlicht, was das Inkrafttreten der Verpflichtungen im Jahr 2017 bedeuten würde.
Auswirkungen auf den Apothekenbetrieb und Haftungsverschärfungen
Setzt man voraus, dass die Kommission das System der Verifizierungsmethodik gemäß der von securPharm gemachten Vorschläge übernimmt, ergeben sich für den Apotheker im Alltag aus dem End-to-end-Prinzip keine weiteren Arbeitsschritte. Sollte das vorgehaltene System den DataMatrix-Code nicht verarbeiten können, kommen insoweit gegebenenfalls finanzielle Belastungen für eine Umstellung auf den Apotheker zu.
Hinsichtlich der Überprüfung des Anti-Manipulationsmerkmals sind für den Apotheker keine gravierenden Auswirkungen zu erwarten; er muss sich bei der Sichtkontrolle lediglich vom Vorhandensein und der Unversehrtheit des Merkmals überzeugen.
Gewichtigere Auswirkungen sind aus haftungsrechtlicher Seite zu erwarten. Bei der Abgabe von Arzneimitteln hat der Apotheker Sorgfaltspflichten zu beachten – insoweit besteht keine Änderung zu der heutigen Situation. Werden jedoch in Zukunft weitere Kontrollmechanismen implementiert, etwa das individuelle Erkennungsmerkmal (Matrix-Code) oder das Anti-Manipulationsmerkmal, so muss der Apotheker vor der Abgabe entsprechende Prüfungen gemäß dem End-to-end-Prinzip oder der Sichtprüfung des Arzneimittels auf Fälschungsverdacht hin vornehmen und haftet für eine sorgfältige Wahrnehmung dieser zusätzlichen Pflichten.
Fazit
Die Auswirkungen der neuen Regelungen auf den Apothekenmarkt sind überschaubar. Schon jetzt werden in aller Regel Apotheker die Arzneimittel vor der Abgabe einscannen. Dass der Computer in Zukunft einen Abgleich zweier Datenbanken vollzieht, bevor er das Arzneimittel zur Abgabe freigibt, bringt – das einwandfreie Funktionieren des gesamten Systems vorausgesetzt – keine spürbare Veränderung mit sich. Die Sichtprüfung des Arzneimittels dürfte in Zukunft eventuell intensiver ausfallen, da anders als heute nicht nur auf oberflächliche Beschädigungen der äußeren Umhüllung, sondern auch auf das Vorhandensein bzw. die Beschädigung von Anti-Manipulationsmerkmalen achtgegeben werden muss.
Mit den erhöhten Pflichten geht der strengere Haftungsmaßstab einher. Um sich nicht dem Vorwurf der Verletzung von Sorgfaltspflichten ausgesetzt zu sehen, sind die genannten Schritte in jedem Fall vor der Abgabe eines Arzneimittels durchzuführen.
Autor
Rechtsanwalt Andreas Frohn, Kanzlei am Ärztehaus, Frehse Mack Vogelsang, Köln, www.kanzlei-am-aerztehaus.de
DAZ 2012, Nr. 43, S. 64



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.