













- DAZ.online
- DAZ / AZ
- DAZ 10/2012
- Zellen außer Kontrolle
Onkologie
Zellen außer Kontrolle
Mechanismen der Krebsentstehung und Eingriffsmöglichkeiten
Tumorzellen entstehen durch Umwandlung normaler Körperzellen. Eine Tumorzelle ist dadurch gekennzeichnet, dass Wachstumskontrollmechanismen bei ihr nicht mehr funktionieren. Als Konsequenz wachsen Tumorzellen autonom und progressiv.
Eine weitere fundamentale Eigenschaft von Krebszellen besteht darin, dass die Zellen, die durch mitotische Teilung aus einer Krebszelle hervorgehen, ebenfalls Tumorzellen sind. Dies zeigt, dass die Eigenschaften, die für den Phänotyp "Krebs" verantwortlich sind, bei der Zellteilung stabil vererbt werden. Obwohl die Möglichkeit besteht, dass man aufgrund bestimmter Erbanlagen ein erhöhtes Risiko besitzt, an Krebs zu erkranken, ist jedoch in aller Regel ein Tumorleiden nicht angeboren. Vielmehr wird die Krankheit irgendwann im Laufe des Lebens erworben. Dazu kommt es, wenn das Genom einer einzigen Zelle der 1014 Zellen eines menschlichen Körpers in ganz bestimmter Weise verändert wird. Wird eine solche Veränderung (Mutation) nicht repariert und wird die mutierte Zelle vom Immunsystem nicht erkannt und eliminiert, hat sich ein Tumor etabliert, der dann zur lebensbedrohlichen Gefahr für den Organismus wird.
Um die Entstehung von Tumoren verstehen zu lernen, muss man sich daher folgende Fragen stellen:
Welche Bereiche des Genoms der betroffenen Zellen sind verändert?
Wie sind diese DNA-Bereiche verändert?
Naheliegend ist, dass die durch Mutationen veränderten DNA-Bereiche entscheidend an der Regulation des Zellwachstums beteiligt sein müssen, denn der Verlust der Wachstumskontrolle ist offensichtlich die eigentliche Ursache für die Entstehung von Krebs.
Das Konzept der Wachstumskontrolle
In einem multizellulären Organismus muss und wird Wachstum sehr subtil und komplex kontrolliert. Anders als in Einzellern wie Bakterien oder Hefen, die klonal wachsen und in denen somit alle Zellen gleich sind, muss in einem multizellulären Organismus, der aus unterschiedlich spezialisierten Zellen besteht, das Wachstum der einzelnen Zellen streng und spezifisch koordiniert werden. Diese Koordination übernehmen zwei ineinandergreifende Kontrollmechanismen:
Wachstumsaktivatoren kontrollieren das Proliferationsvermögen einer Zelle positiv. Sie sind aktiviert, wenn eine Mitose, d. h. eine Zellteilung, eingeleitet wird.
Wachstumsinhibitoren kontrollieren das Proliferationsvermögen einer Zelle negativ. Sie sind aktiviert, wenn die Zellteilung unterbunden werden muss.
Kontrolle bedeutet dabei immer, dass der zu kontrollierende gewisse Prozess bei Bedarf entweder an- oder abgeschaltet werden kann. Dies trifft auch – und in besonderem Maße – für die Wachstumskontrolle zu. Wachstumskontrolle geht gleichermaßen dann verloren, wenn Wachstumsaktivatoren nicht mehr inaktiviert, oder wenn Wachstumsinhibitoren nicht mehr aktiviert werden können. Meist ist in einer Tumorzelle beides realisiert, denn Krebsentstehung ist ein multifaktorielles Ereignis.
Damit lassen sich auch die beiden gestellten Fragen beantworten:
Bei der Entstehung von Tumoren sind DNA-Bereiche mit Genen betroffen, die die Informationen für Wachstumsaktivatoren und für Wachstumsinhibitoren kodieren. Diejenigen Gene für Wachstumsaktivatoren bezeichnet man als Proto-Onkogene, die Gene für Wachstumsinhibitoren werden Tumor-Suppressorgene genannt.
Mutationen in diesen DNA-Bereichen haben unterschiedliche Konsequenzen je nachdem, ob es sich bei dem mutierten Gen um ein Proto-Onkogen oder um ein Tumorsuppressorgen handelt. Mutationen in Proto-Onkogenen führen in der Regel dazu, dass die Produkte der Proto-Onkogene, also die Wachstumsaktivatoren, nicht mehr abgeschaltet werden können und permanent Proliferationssignale aussenden. Mutationen in einem Tumor-Suppressorgen hingegen führen in der Regel dazu, dass das Gen inaktiviert wird, d.h. dass gar kein funktionsfähiger Wachstumsinhibitor mehr gebildet wird.
Onkogene
Mit molekularbiologischen Methoden gelang es, etliche der für die Tumorentstehung verantwortlichen Gene zu identifizieren. Es konnte gezeigt werden, dass sich durch DNA aus Tumoren die Zellen der Mäuse-Zelllinie NIH/3T3-Zellen maligne transformieren lassen. Diese Mäuse-Zelllinie besitzt die Eigenschaft, permanent zu wachsen, zeigt aber sonst keine weiteren Merkmale einer Krebszelle. Schleust man Tumor-DNA in diese Zellen ein, entwickeln sich aus den so modifizierten Zellen Tumorzellen. Dies zeigt sich u. a.
durch eine veränderte Morphologie der Zellen,
durch Verlust der Kontaktinhibition und damit der Fähigkeit, in einer Zellkulturplatte nicht nur bis zu einer geschlossenen Zellschicht zu wachsen zu können, sondern darüber hinaus auch Zellhaufen bilden zu können,
durch die erworbene Fähigkeit, in Nackt-Mäusen als solide Tumoren wachsen zu können. Bei Nackt-Mäusen handelt es sich um einen Mäusestamm, der durch eine Mutation nur noch sehr wenig Thymusgewebe bildet. Diesen Mäusen fehlt daher das zelluläre Immunsystem (das T-Zell-Immunsystem), das bei der Abwehr von Tumorzellen von großer Bedeutung ist.
Aus transformierten NIH/3T3-Zellen kann man wiederum DNA isolieren, diese erneut in NIH/3T3-Zellen einschleusen, und man erhält wieder Tumorzellen. Offensichtlich ist also die Fähigkeit, eine Zelle maligne zu transformieren, übertragbar und stabil. Erforderlich ist hierfür ausschließlich die DNA – also das Erbmaterial – einer Tumorzelle.
Diese Entdeckung war sensationell, denn nun realisierte man, dass in jeder Zelle unseres Körpers die Basisinformation zur Transformation von Zellen vorhanden ist. Onkogene – wie die Informationseinheiten, die Tumoren erzeugen können, fortan genannt wurden – sind mutierte Varianten sogenannter Proto-Onkogene. Proto-Onkogene sind essenzielle Gene, deren Produkte regulatorische Funktionen in Wachstums-, Differenzierungs- und Entwicklungsprozessen normaler Zellen wahrnehmen. Inaktiviert man die Proto-Onkogene, so können die Zellen nicht überleben; verändert man die Proto-Onkogene in definierter Weise, so werden sie zu Onkogenen, die dafür verantwortlich sind, dass die Zelle ihre Wachstumskontrolle verliert.
Bei den Veränderungen, die dazu führen, dass ein Proto-Onkogen in ein Onkogen umgewandelt wird, handelt es sich immer um dominante Mutationen. Das bedeutet, dass bereits die Veränderung eines der beiden auf den homologen Chromosomen einer diploiden Zelle vorhandenen Proto-Onkogene ausreicht, um den Anstoß zur malignen Entartung der Zelle zu geben.
Onkogene entstehen aus den Proto-Onkogenen
- durch Änderung einer einzelnen Base im Gen selbst (Punktmutation),
- durch Neukombination zweier Gene (Translokation),
- durch lokale Vermehrung des Proto-Onkogens (Amplifikation),
- durch Mutationen im Kontrollbereich des Proto-Onkogens (Aktivierung).
Bisher konnten mehr als 230 Proto-Onkogene identifiziert und charakterisiert werden. Die als molekulare "Schalter" fungierenden Produkte dieser Proto-Onkogene sind Teil eines komplexen Informations- und Signalnetzes einer Zelle. Sie übermitteln Proliferationssignale aus der Zellumgebung oder aus dem Zytoplasma der Zelle in den Zellkern. Als Konsequenz werden dann Proteine gebildet, die die Teilung der Zelle einleiten. Das koordinierte Zusammenspiel dieser wichtigen Faktoren garantiert letztlich das kontrollierte Wachstum einer Zelle im komplexen Gewebeverband eines Organismus. Wird dieses System gestört, kommt es zur malignen Entartung, die gekennzeichnet ist durch unkontrolliertes, progressives und invasives Wachstum der sich stetig vermehrenden Zellen.
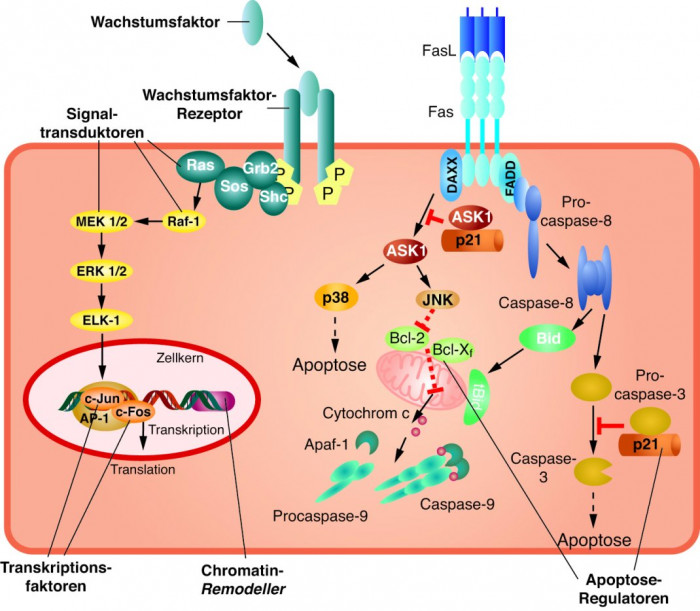
Proto-Onkogene lassen sich nach der Funktion ihrer Produkte klassifizieren (Abb. 1). Bei den Produkten handelt es sich immer um Proteine. Es sind:
Transkriptionsfaktoren,
- Chromatin-Remodeller,
- Wachstumsfaktoren,
- Wachstumsfaktor-Rezeptoren,
- Signaltransduktoren und
- Apoptose-Regulatoren.
Transkriptionsfaktoren. In der ersten Klasse sind Proteine zusammengefasst, die im Kern lokalisiert sind. Vielfach handelt es sich um Transkriptionsfaktoren, die direkt an der Expressionskontrolle der genetischen Information der Zelle beteiligt sind. Um ihre Funktion zu entfalten, interagieren diese Proteine oft mit anderen Proteinen. So dimerisiert beispielsweise Fos mit Jun, woraus dann der Transkriptionsfaktor AP1 resultiert. Dieser kontrolliert eine ganze Schar von Genen, die in der Zellteilung eine wichtige Rolle spielen.
Solche Transkriptionsfaktoren, deren Aktivitäten natürlich streng kontrolliert sein müssen, können durch bestimmte Mutationen ihre Kontrolle zugunsten einer konstitutiven Aktivierung verlieren. Eine Möglichkeit in diese Richtung sind chromosomale Translokationen. Diese beobachtet man häufig in lymphoiden Tumoren und gelegentlich auch in soliden Tumoren. Manche Tumoren sind durch solche Translokationen regelrecht definiert.
Chromatin-Remodeller. Die Proteine der zweiten Klasse, die Chromatin-Remodeller, modifizieren die Struktur des Chromatins, die wiederum entscheidenden Einfluss auf die Kontrolle der Genexpression, der Replikation und der Genreparatur nimmt. Zwei verschiedene Enzymtypen sind am Chromatin-Remodelling beteiligt:
- ATP-abhängige Enzyme, die die Nukleosomen neu positionieren, und
- Enzyme, die die N-terminalen Enden bestimmter Histone modifizieren.
Das Muster der Histonmodifikationen ist die Basis für den epigenetischen Code, der die Interaktionen zwischen den Nukleosomen und den Chromatin-assoziierten Proteinen kontrolliert. Diese Interaktionen wiederum bestimmen die Struktur des Chromatins und der Transkriptionskompetenz des jeweiligen Chromosomenabschnitts.
Beispielsweise ist das ALL1 (MLL)-Genprodukt Teil eines sehr großen, stabilen Multiproteinkomplexes. Etliche Proteine dieses Komplexes sind Transkriptionsfaktoren, andere Histon-Methylasen oder RNA-Prozessierungsenzyme. Der Gesamtkomplex remodelliert, acetyliert, deacetyliert und methyliert Nukleosomen und freie Histone. Dieses wichtige und komplexe Netzwerk gerät durcheinander, wenn ALL1 in Translokationen involviert wird. Das ist sehr häufig bei der akuten lymphatischen Leukämie (ALL) und der akuten myeloischen Leukämie (AML) der Fall. In über 80% dieser Leukämien findet man Fusionen des ALL1 (MLL)-Gens mit einem von über 50 Partnergenen.
Wachstumsfaktoren. In die dritte Klasse fallen sezernierte Proteine, die entweder als Wachstumsfaktoren identifiziert sind oder die aufgrund struktureller Homologien der Gruppe der Wachstumsfaktoren zuzuordnen sind. Werden Wachstumsfaktoren konstitutiv exprimiert, so führt dies zu fatalen Fehlregulationen. Der Plättchenwachstumsfaktor (PDGF) mit seiner α- und β-Untereinheit spielt während der Blutgerinnung eine wichtige Rolle. Er induziert die Proliferation verschiedener Zellen und stimuliert Fibroblasten für die Wundheilung. Nun besitzt beispielsweise das sis -Onkogen-Produkt eine große Homologie zur PDGF-β-Kette. Wird das sis-Onkogen überexprimiert, induziert dies die Transformation von Fibroblasten, die einen PDGF-Rezeptor exprimieren.
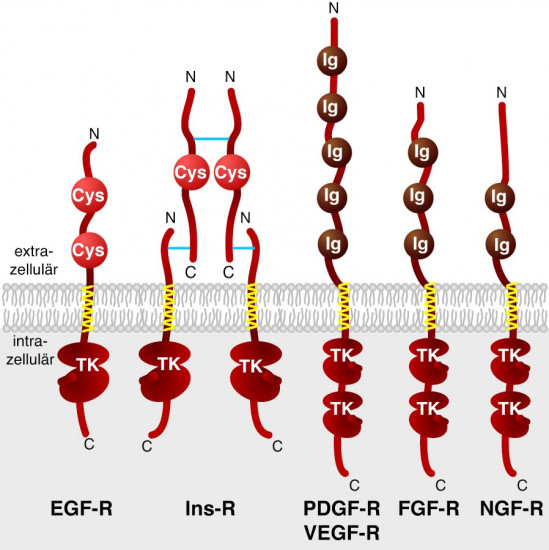
Wachstumsfaktor-Rezeptoren. In der vierten Gruppe finden sich mutierte Varianten bereits charakterisierter Zell-Oberflächenrezeptoren. So ist ERB-B eine Variante des epidermalen Wachstumsfaktor-Rezeptors [EGF-Rezeptor] und FMS entspricht dem Rezeptor des colony-stimulating factor 1 [CSF-1]. In dieser Gruppe sind aber auch Genprodukte angesiedelt, die strukturell den Oberflächen-Rezeptoren zuzuordnen sind, wobei jedoch die spezifischen Liganden noch nicht bekannt sind (z. B. die Produkte der Proto-Onkogene trk, met, neu und ros). Meist besitzen diese Proteine eine Protein-Tyrosin-Kinaseaktivität, d. h. sie modifizieren bestimmte Zielproteine an Tyrosinresten mit Phosphatgruppen (Abb. 2). Normalerweise wird die Aktivität dieser Proteine subtil reguliert. Das transformierende Potenzial der Onkogene rührt wohl daher, dass deren Produkte – im Gegensatz zu den Produkten der Proto-Onkogene – permanent in einem aktivierten Zustand vorliegen, auch dann, wenn der physiologisch aktivierende Stimulus fehlt.
Signaltransduktoren. In der fünften Gruppe sind Onkogene zusammengefasst, die für Komponenten von Signaltransduktionswegen kodieren. Sie lassen sich in zwei Gruppen aufteilen:
- Nicht-Rezeptorkinasen
- – Tyrosin-Kinasen (z. B. ABL, LCK, SRC)
- – Serin/Threonin-Kinasen (z. B. AKT, RAF1, MOS, PIM1)
- Guanosintriphosphat-bindende Proteine (G-Proteine)
Die Nicht-Rezeptorkinasen werden dann zu Onkoproteinen, wenn sie konstitutiv, d. h. permanent und unkontrolliert, als Protein-Tyrosin- oder als Serin/Threonin-Kinasen wirken.
Das wichtigste Guanosintriphosphat-bindende Protein unter den Proto-Onkogenprodukten ist das RAS-Protein, das GTP nicht nur bindet, sondern auch hydrolysiert. Distinkte Punktmutationen machen aus dem Proto-Onkogen das Onkogen. Funktionell unterscheiden sich die Produkte der Onkogene von den Produkten der nicht-mutierten Gene dadurch, dass sie eine deutlich schlechtere GTP-spaltende Aktivität (GTPase-Aktivität) besitzen.
Apoptose-Regulatoren. Die sechste Gruppe enthält die Apoptose-Regulatoren. Das Bcl-2-Gen, das bei der Initiation fast aller follikulärer Lymphome und einiger diffuser B-Zell-Lymphome beteiligt ist, kodiert ein zytoplasmatisches Protein, das ins Mitochondrium einwandern kann und dadurch das Absterben von Zellen verhindert. In bestimmten Tumoren ist Bcl-2 daher, ebenso wie der enge Verwandte Bcl-xL, hochreguliert.
Diese Hochregulation wirkt dem in manchen Situationen so wichtigen Zelltod entgegen, denn Apoptose wird u. a. durch die Inaktivierung von Bcl-2 durch Proteine induziert, die eine BH3-Domäne (Bcl-2-Homologiedomäne-3) besitzen. Diese Inaktivierung stößt einen Signalweg an, der schließlich Caspasen aktiviert, die direkt das apoptotische Geschehen kontrollieren.
Biochemische Wirkmechanismen der Proto-Onkogene
Obwohl die Zahl der verschiedenen Proto-Onkogene bereits relativ groß ist, lassen sich vereinfacht deren Wirkungen auf nur drei unterschiedliche biochemische Mechanismen reduzieren.
Der erste Mechanismus ist eine Protein-Phosphorylierung, wobei Serin-, Threonin- und Tyrosin-Reste in Proteinen als Substrate dienen können. Protein-Serin/Threonin-Kinasen waren bereits seit Langem bekannt, während Protein-Tyrosin-Kinasen erst durch das Studium der Proto-Onkogene entdeckt wurden. Letztere können wiederum in drei Klassen unterteilt werden, je nachdem ob sie in der Plasmamembran verankert sind (z. B. der Epidermale Wachstumsfaktor-Rezeptor [EGF-Rezeptor]), ob sie im Zytoplasma gelöst bzw. Membran-gebunden vorkommen (z. B. das SRC-Protein), oder ob sie im Kern lokalisiert sind (z. B. das ABL-Genprodukt). Zu den Protein-Serin/Threonin-Kinasen, die an der Zellproliferation kontrollierend beteiligt sind, ist neben den vier Onkogenprodukten AKT, RAF1, MOS, PIM1 auch noch die Protein-Kinase C zu zählen. Dieses Enzym wird u. a. auch durch Tumorpromotoren wie Phorbolester aktiviert.
Der zweite Mechanismus beruht auf der Transmission von Signalen durch die Hydrolyse von GTP (GTPase-Aktivität). Hier ist das ras-Onkogen als markanter Vertreter zu nennen. In diese Klasse fallen aber auch die heterotrimeren G-Proteine, die aus den drei Untereinheiten α, β und γ zusammengesetzt sind. In der Regel ist die α-Untereinheit des Komplexes, die auch für die GTP-Bindung und -Hydrolyse verantwortlich ist, mutiert. Die entsprechenden Onkogenprodukte werden als GS (stimulatory G proteins) oder als GI (inhibitory G proteins) bezeichnet. Dass mutierte ras-Gene (also ras-Onkogene) eine zentrale Rolle bei Tumoren spielen, kann u. a. auch daran gezeigt werden, dass Antikörper, die die ras-Genprodukte binden, die mitogene Wirkung bestimmter Wachstumsfaktoren und das transformierende Potenzial bestimmter Onkogene neutralisieren können. Mutationen in ras werden in nahezu 30% aller Tumoren gefunden.
Der dritte Mechanismus beruht auf der Transkriptionskontrolle, d. h. auf dem Bereich, der für die zeitlich und räumlich differenzierte Realisierung der genetischen Information verantwortlich ist. Hier sind fos und myc als bekannte Proto-Onkogene zu nennen, deren Produkte nicht nur als Transkriptionsfaktoren sondern teilweise auch als Replikationsfaktoren fungieren können. Einige dieser Faktoren sind erst aufgrund von Onkogenstudien entdeckt worden.
Tumor-Suppressorgene
Als Tumor-Suppressorgene wurden bereits weiter oben Gene definiert, deren Produkte an der Verhinderung der Zellproliferation beteiligt sind. Man bezeichnet diese Gene auch als Wachstums-Suppressorgene, rezessive Onkogene oder Anti-Onkogene.
Das Wissen um diese Komponenten in dem komplexen Geschehen der Tumorentstehung ist wesentlich jünger. Das hat natürlich Gründe. Aus heutiger Sicht war die Arbeit mit Onkogenen einfach, da es sich bei der Umwandlung von Proto-Onkogenen zu Onkogenen um dominante Mutationen handelte. Das Einschleusen eines Onkogens allein verursachte die maligne Transformation bestimmter Zellen, z. B. der NIH3T3-Zellen.
Beobachtungen von Zellbiologen hingegen zeigten ganz andere Effekte, die man als das Fusionsparadoxon umschreiben kann. Danach dominiert normales Wachstum über maligne Entartung. Man beobachtete nämlich, dass bei der Fusion einer normalen Zelle und einer Tumorzelle die Hybridzelle nicht etwa die Eigenschaften einer Tumorzelle erworben hatte, sondern dass die Hybridzelle sich wachstumskontrolliert verhielt. Demnach müssen normale Zellen über Gene verfügen, die über die Onkogene dominieren und deren Produkte unkontrolliertes Wachstum verhindern. Tumorzellen hingegen scheint diese Information zu fehlen.
Heute weiß man, dass die Information, die offensichtlich den Tumorzellen fehlt, die Information ist, die durch Tumor-Suppressorgene kodiert wird. Die Schwierigkeit, Tumor-Suppressorgene zu identifizieren besteht darin, dass sie für die Tumorentstehung erst dann Relevanz erlangen, wenn beide Kopien (= Allele) auf den beiden homologen Chromosomen einer diploiden Zelle defekt sind. Das leuchtet ein, wenn man sich vorstellt, dass die Produkte dieser Gene an der Produktion oder an der Vermittlung negativer, zytostatischer Faktoren beteiligt sind. Selbst wenn eins der beiden Allele eines Tumor-Suppressorgens defekt ist, stellt das verbliebene intakte Allel in der Regel noch so viel Produkt bereit, dass eine Proliferationskontrolle gewährleistet bleibt. Erst wenn auch das zweite Allel durch Mutation inaktiv ist, kommt es zum Kontrollverlust und möglicherweise zur unkontrollierten Proliferation. Die Inaktivierung nur eines Allels eines Tumor-Suppressor-Genpaares in einer diploiden Zelle ist daher rezessiv, weshalb man die Gene auch als rezessive Onkogene bezeichnet.
Tumor-Suppressorgene wurden entdeckt durch das konsequente Studium bestimmter Tumorerkrankungen, die sehr häufig bereits im Kindesalter auftreten, wie beispielsweise das Retinoblastom oder der Wilms-Tumor. Diese Krankheiten standen bereits seit Langem im Verdacht, eine vererbbare Komponente zu enthalten, da diese Tumoren in bestimmten Familien gehäuft auftreten. Hier nimmt Krebs tatsächlich – zumindest prinzipiell – den Charakter einer Erbkrankheit an, was auch einleuchtet, denn damit ein Tumor-Suppressorgen sein onkogenes Potenzial entfalten kann, müssen beide Allele des Gens inaktiv sein. Die Wahrscheinlichkeit, dass dieser Fall eintritt, ist deutlich erhöht, wenn schon bei Geburt eines der beiden Allele defekt ist. Menschen, die bezüglich eines Tumor-Suppressorgen-Lokus‘ heterozygot sind, tragen daher ein wesentlich größeres Krebsrisiko, so dass es oft bereits im Kindesalter zu Tumoren kommt.
Biochemisch betrachtet fungieren die Produkte von Tumor-Suppressorgenen als Vermittler von Antiproliferationssignalen. Biologisch gesehen sind sie Teil eines komplexen Systems, das Zellteilung stoppt. Dies geschieht transient durch Anhalten des Zellzyklus, programmiert durch Einleitung der Differenzierung oder terminal durch Kanalisierung der Physiologie einer Zelle in Richtung Alterung und Tod. Der funktionelle Verlust dieser Gene kehrt alle diese Prozesse um, so dass zwar die einzelne Zelle Unsterblichkeit erlangt, dies aber letztlich als Konsequenz nach sich zieht, dass ein ganzer Organismus an der unkontrollierten Expansion einer einzelnen Zelle zugrunde geht.
Krebs wird jedoch niemals durch ein einzelnes Ereignis verursacht. In der Regel sind Tumorzellen ganz massiv genetisch geschädigt, d. h. man findet mehrere aktivierte Onkogene und meist auch den kompletten Verlust von mindestens einem der bekannten Tumor-Suppressorgene. Offensichtlich sind also viele Schritte erforderlich, um schließlich eine vitale Tumorzelle zu etablieren.
Praktische Konsequenzen der molekularen Tumorforschung
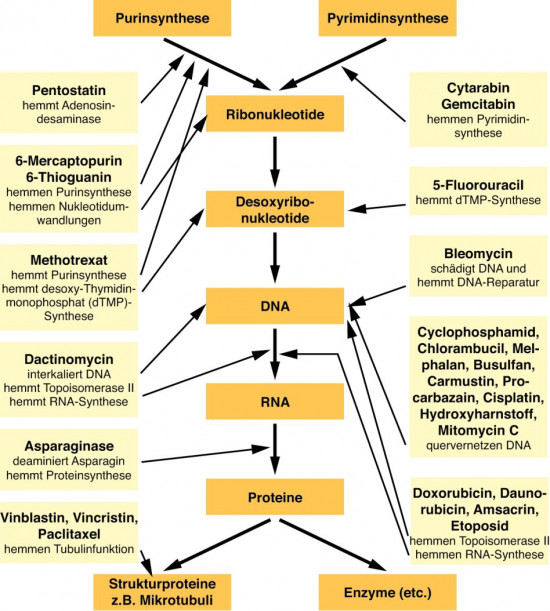
Längst haben diese Erkenntnisse der modernen Tumorforschung zu neuen Ansätzen bei der Behandlung von Tumorerkrankungen geführt. Waren bis vor einigen Jahren noch nahezu alle Strategien der Tumortherapie darauf ausgelegt, den Tumor zu eliminieren (Abb. 3), ließen sich auf der Basis des Wissens um die molekularen Mechanismen der Tumorentstehung neue Konzepte erarbeiten. Die drei Säulen der Tumorbehandlung, Chirurgie, Bestrahlung, Chemotherapie, wurden und werden weiter ergänzt durch medikamentöse Behandlungsprinzipien, die nicht nur zum Ziel haben, den Tumor zu eliminieren, sondern mit deren Hilfe die Wachstumskontrolle in einer Tumorzelle wiederhergestellt wird. Dies gelingt z. B. durch Inhibitoren, die aktivierte Onkogenprodukte inaktivieren oder die die Aufgabe von Tumor-Suppressorgenprodukten übernehmen, die durch Mutationen verloren gegangen sind.
Letztlich wird immer deutlicher, dass mit einer Heilung einer Tumorerkrankung nur gerechnet werden kann, wenn das eigene Immunsystem den Tumor eliminiert. Daraus folgen zwei Strategien:
Es sind Maßnahmen zu treffen, die das unkontrollierte Tumorwachstum stoppen und die den Tumor in seiner Größe so weit wie möglich verkleinern.
Es sind Maßnahmen zu treffen, die das Immunsystem stärken, um es in die Lage zu versetzen, Rest-Tumorzellen aufzuspüren und zu eliminieren.
In den Maßnahmenkatalog der ersten Gruppe fallen natürlich die klassischen Zytostatika (Abb. 3). Hierbei handelt es sich zunächst einmal um "ordinäre Zellgifte", die eine selektive Toxizität vor allem dadurch entfalten, dass sich die zytotoxische Wirkung auf eine schnell wachsende Krebszellpopulation viel stärker auswirkt, als auf eine langsam oder gar nicht wachsende Normalzellpopulation. Aus diesem Grund wirken diese Wirkstoffe auch bei schnell proliferierenden Tumoren besonders gut, während sie bei langsam wachsenden Tumoren kaum wirksam sind.
In die erste Gruppe fallen aber auch viele neuartige Wirkstoffe, die sehr gezielt die Tumor-treibenden Onkoproteine ansteuern und so ihre biologischen Effekte neutralisieren. Hier lautet ein neues Paradigma "Stoppen statt Töten". Das zugrunde liegende Prinzip ist ein antagonistisches Prinzip.
Hindert beispielsweise ein Antikörper einen deregulierten Wachstumsfaktor daran, seinen spezifischen Rezeptor durch Bindung zu stimulieren, wird der Tumor in einen Wachstumsstillstand gedrängt, aber zunächst einmal nicht eliminiert. Entsprechende Wirkstoffe und ihre Zielstrukturen sind in Tabelle 1 aufgeführt
Tab. 1: Rekombinante Antikörper, die Signaltransduktionskaskaden unterbrechen und ihre Zielstrukturen | ||
Handelsname |
Wirkstoff |
Zielstruktur |
Erbitux®
|
Cetuximab |
EGFR |
Vectibix®
|
Panitumumab |
EGFR |
Herceptin®
|
Trastuzumab |
HER2/neu |
Avastin®
|
Bevacizumab |
VEGF |
Gleiches gilt für niedermolekulare Tyrosin-Kinase-Inhibitoren (Tab. 2), die das konstitutive Feuern der mutierten Rezeptoren unterbrechen. Als Konsequenz dieses Mechanismus müssen diese Wirkstoffe permanent in ausreichend hohen Dosen vorhanden sein. Denn sobald man die Medikation absetzt, beginnen die Tumorzellen wieder zu wachsen.
Tab. 2: Tyrosin-Kinase-Inhibitoren und deren Spezifität | |||||||
Wirkstoff |
Bcr-Abl |
EGFR |
HER2 |
PDGFR |
VEGFR |
cKit |
Raf |
Dasatinib |
X |
X |
X |
||||
Imatinib |
X |
X |
X |
||||
Nilotinib |
X |
X |
X |
||||
Sorafenib |
X |
X |
X |
X |
|||
Sunitinib |
X |
X |
X |
||||
Erlotinib |
X |
||||||
Gefitinib |
X |
||||||
Lapatinib |
X |
X |
|||||
Einen Übergang in das zweite Prinzip, das auf der Stärkung des eigenen Immunsystems beruht, bilden die rekombinanten Antikörper, die an Zelloberflächenproteine binden (Tab. 3).
Tab. 3: "Onkolytische Antikörper" und ihre Zielstrukturen | ||
Handelsname |
Wirkstoff |
Zielstruktur |
Erbitux®
|
Cetuximab |
EGFR |
Vectibix®
|
Panitumumab |
EGFR |
Herceptin®
|
Trastuzumab |
HER2/neu |
MabCampath®
|
Alemtuzumab |
CD52 |
MabThera®
|
Rituximab |
CD20 |
Arzerra®
|
Ofatumumab |
CD20 |
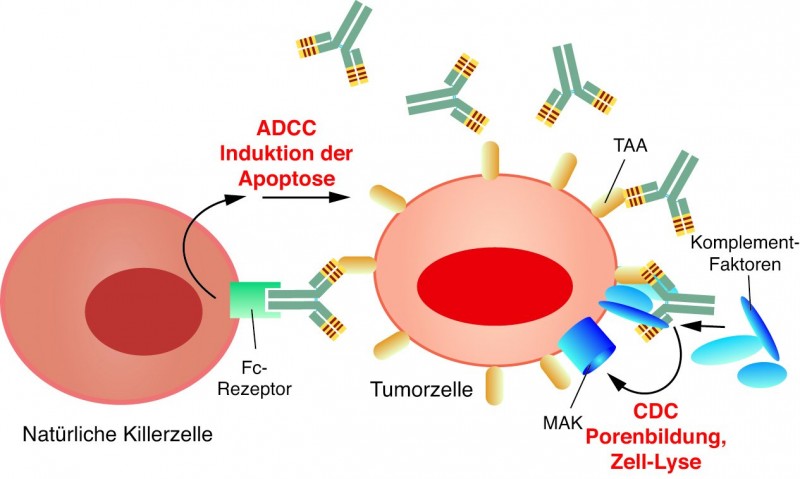
Hier kommen agonistische Ansätze zum Tragen, indem das Immunsystem als solches aktiviert wird. Durch ihre Bindung an Tumorzellen werden zwei Tötungsmechanismen aktiviert, die als Antikörper-abhängige zelluläre Zytotoxizität (ADCC) oder als Komplement-abhängige Zytotoxizität (CDC) bezeichnet werden (Abb. 4). Bei der ADCC binden NK-Zellen und auch andere Zellen, die einen Fc-Rezeptor exprimieren, an die Effektorregion des Antikörpers. Die Zellen werden dadurch aktiviert und setzen den Inhalt von Granula bzw. Vesikeln (Perforin, Granzyme, lytische Enzyme, TNF) frei und töten so die opsonisierten Zellen ab. Bei der CDC wird durch den Immunkomplex aus Antikörper und Tumorzelle das Komplement-System aktiviert, das in seiner Endstrecke einen Membran-angreifenden Komplex (MAK) ausbildet, der die Zellmembran durchlöchert und die Tumorzelle so in die Apoptose schickt.
Abb. 4: Exprimieren Tumorzellen spezifische Antigene
(Tumor-assoziiertes Antigen, TAA), können sehr effizient Antikörper eingesetzt werden, die selektiv an dieses Antigen binden. Durch die Antikörperbindung wird einerseits die Antikörper-abhängige zelluläre Zytotoxizität (ADCC) über beispielsweise Natürliche Killerzellen aktiviert (links). Zum anderen können Komplementfaktoren mit dem Immunkomplex auf der Tumorzelle assoziieren und einen Membran-angreifenden Komplex (MAK) ausbilden, wodurch die Zelle lysiert (CDC).
Grafik: Zündorf
Im Bereich dieser zweiten Option werden Ansätze zunehmend bedeutender, die darin bestehen, das Immunsystem generell zu stärken, um es in die Lage zu versetzen, Rest-Tumorzellen aufzuspüren und zu eliminieren.
Wirkstoffe, die hier zugelassen sind, sind beispielsweise Interleukin 2 (Proleukin®), Tumornekrosefaktor alfa (Beromun®) oder Interferon alfa (Roferon® und Intron® A). Die Substanzen haben starke Nebenwirkungen, und oft sind die Responderraten sehr klein (bei Proleukin® beispielsweise nur 15%). Wer allerdings auf die Medikamente anspricht, kann mit ausgezeichneten Ergebnissen rechnen.
Um nicht die hochaktiven Biomoleküle selbst applizieren zu müssen und dadurch starke systemische Reaktionen zu provozieren, versucht man nun, um dem Tumor herum ein immunaktives Milieu zu etablieren.
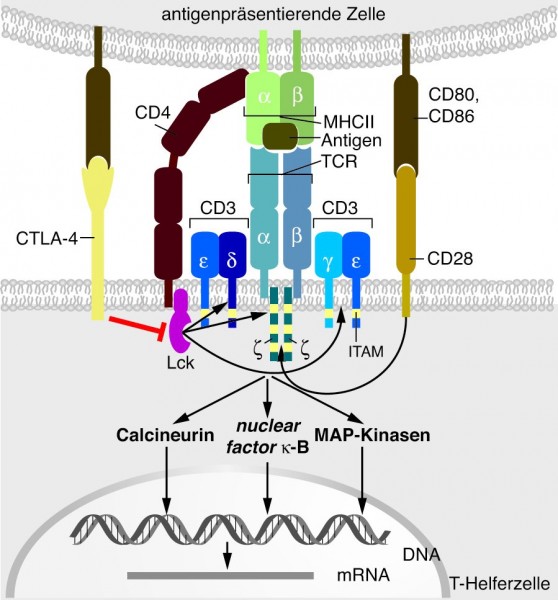
Ein solcher Wirkstoff ist beispielsweise Ipilimumab (Yervoy®), ein vollständig humaner Antikörper, der gegen das Protein CTLA-4 (Cytotoxic T-Lymphocyte Antigen-4) gerichtet ist. CTLA4, das auch als CD152 bekannt ist, konkurriert mit CD28 um Bindung an die Liganden CD80 und CD86, wobei es eine höhere Affinität besitzt als CD28 (Abb. 5). CD152-Signale inhibieren die Signaltransduktion des T-Zellrezeptors und schwächen so die Aktivität von T-Zellen ab. Außerdem führen CD152-Signale zur Induktion der Expression von TGF-β, einem Zytokin, das Immunreaktionen unterdrückt. Aus diesen Zusammenhängen erschließt sich nun die Logik einer Intervention mit Ipilimumab. Denn Ipilimumab neutralisiert die Funktion von CD152 (CTLA-4) und damit das immunsupprimierende Potenzial dieses Faktors. Yervoy® ist zur Behandlung von fortgeschrittenen (nicht resezierbaren oder metastasierten) Melanomen bei Erwachsenen indiziert, die bereits zuvor eine Therapie erhalten haben.
Abb. 5: Bei der Aktivierung von Antigen-spezifischen T-Zellen
interagieren die antigenpräsentierenden Zellen (APC) neben der Wechselwirkung zwischen dem MHCII-Antigen-Komplex und dem T-Zell-Rezeptor (TCR) noch über andere Oberflächenmoleküle mit T-Zellen. B7 (CD80 bzw. CD86) auf der APC kann entweder an CD28 oder an CTLA-4 (CD152) auf T-Zellen binden. Während jedoch die Bindung an CD28 ein kostimulatorisches Signal für die T-Zell-Aktivierung darstellt, führt die Bindung an CTLA-4 zu einer Inaktivierung der T-Zelle.
Grafik: Zündorf
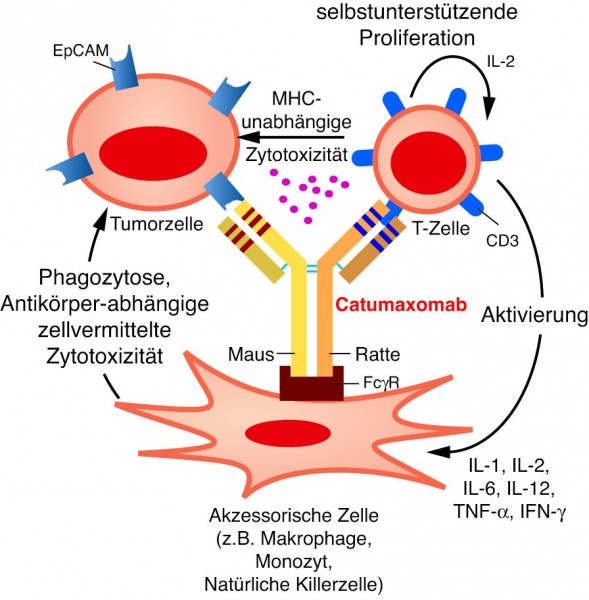
Ein weiteres Prinzip in diese Richtung wird mit Catumaxomab (Removab®) aufgezeigt. Catumaxomab gehört zur Triomab®-Plattform der Münchner Firma TRION Pharma und ist ein bispezifischer, trifunktionaler Ratte/Maus-Hybrid-Antikörper, der über die klassische Hybridom-Technologie hergestellt wurde und aus einer Ratte/Maus-Hybrid-Hybridomzelllinie gewonnen wird. Zunächst wurde eine murine Hybridomazelllinie hergestellt, die den IgG2a-Antikörper gegen EpCAM produziert. Eine Zelllinie aus der Ratte liefert den IgG2b-Antikörper gegen CD3. Diese beiden Hybridomzelllinien wurden wiederum zu einer Ratte/Maus-Hybrid-Hybridomzelllinie fusioniert, die nun unter anderem auch den gewünschten Hybridantikörper sezerniert, der zur Hälfte aus dem Maus-Antikörper und zur Hälfte aus dem Ratten-Antikörper besteht (Abb. 6). Dieser Hybridantikörper ist nun in der Lage, mit einer variablen Domäne EpCAM und mit der anderen CD3 zu binden. Während EpCAM praktisch auf allen Karzinomzellen exprimiert ist, ist CD3 mit dem T-Zellrezeptor assoziiert und deshalb auf allen T-Zellen zu finden.
Abb. 6: Catumaxomab
ist ein Ratte-Maus-Hybridantikörper, der von einer Ratte-Maus-Hybrid-Hybridomzelllinie sezerniert wird. Der Ratten-Anteil des Antikörpers bindet an CD3 auf T-Zellen, während der Maus-Anteil Ep-CAM erkennt. Dadurch werden T-Zellen in die Nähe der adressierten Tumorzelle gebracht und aktiviert. Das führt dazu, dass die Tumorzelle über Perforine und Granzym B abgetötet wird. Zusätzlich kann Catumaxomab über den konstanten Fc-Teil akzessorische Immunzellen wie Makrophagen oder Natürliche Killerzellen mobilisieren, die dann ebenfalls gebundene Tumorzellen abtöten.
Grafik: Zündorf
Der Wirkmechanismus von Catumaxomab liegt zum einen darin, dass T-Zellen an die EpCAM-exprimierenden Tumorzellen rekrutiert werden und diese dann zerstören; zum anderen soll der konstante Teil des Antikörpers akzessorische Zellen wie Monozyten oder Makrophagen dazu bringen, den Komplex aus Antikörper und Tumorzelle zu eliminieren. Damit wirkt Catumaxomab als quasi katalytischer Antikörper.
Removab® ist indiziert zur intraperitonealen Behandlung des malignen Aszites bei Patienten mit EpCAM-positiven Karzinomen, für die keine Standardtherapie zur Verfügung steht oder bei denen diese nicht mehr anwendbar ist. Dies ist zwar eine Nischen-Indikation. Das Prinzip ist allerdings äußerst plausibel, so dass mit weiteren Antikörpern dieses Typs und weiteren Indikationen gerechnet werden kann.
Fazit
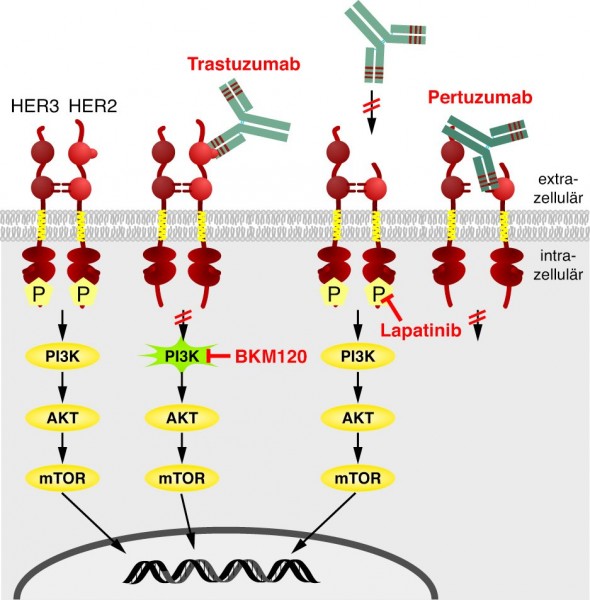
Noch fehlt es an durchschlagenden Erfolgen bei der Tumorbehandlung. Wer von einem Tumorleiden geheilt wird, hat noch Glück gehabt. Allerdings zeigen die sehr rationalen Ansätze, die auf einem tiefgreifenden molekularen Verständnis der Tumorbiologie beruhen, immer größere Erfolge. Beispielsweise versucht man inzwischen, die Resistenz von Mammakarzinomzellen gegen eine Behandlung mit Trastuzumab durch eine Kombination aus verschiedenen Wirkstoffen zu umgehen, die an unterschiedlichen Zielstrukturen innerhalb der Signaltransduktionskaskade angreifen (Abb. 7).
Abb. 7: Beim Mammakarzinom
findet sich häufig eine Überexpression des HER2-Rezeptors, wodurch permanent ein Wachstumssignal weitergegeben wird. Zwischengeschaltet sind beispielsweise die Signalmediatoren PI3K, AKT und mTOR. Bindet der Antikörper Trastuzumab an HER2, kann der Rezeptor kein Signal mehr weiterleiten. Manche Mammakarzinome reagieren trotz HER2-Überexpression nicht auf eine Behandlung mit Trastuzumab. Gründe dafür können beispielsweise sein, dass ein nachgeschalteter Signalmediator mutiert ist und nicht mehr abgeschaltet werden kann (hier als Beispiel PI3K), oder dass die extrazelluläre Domäne, an die Trastuzumab bindet, nicht mehr vorhanden ist. Alternative Therapieansätze könnten dann der Antikörper Pertuzumab sein, der an der Domäne bindet, die zur Dimerisierung des HER2-Rezeptors mit einem anderen Rezeptormolekül nötig ist, oder die Kinase-Inhibitoren Lapatinib und BKM120 oder auch eine Kombination aus den verschiedenen Molekülen.
Grafik: Zündorf
Ein Problem, das es zu lösen gilt, liegt in der großen genetischen Heterogenität besonders von fortgeschrittenen Tumoren. Hier gilt es, die molekulare Diagnostik noch stärker auszubauen und einzusetzen, um die Zielstrukturen zu identifizieren, die einen Tumor hinsichtlich seines Proliferationsverhaltens treiben. Gelingt das, werden auch sehr schnell die Responderraten größer, und eine Tumorbehandlung wandelt sich von einem empirischen "trial and error"-Ansatz zu einer hoch rationalen Intervention.
Autoren
Prof. Dr. Theo Dingermann
Dr. Ilse Zündorf
Institut für Pharmazeutische Biologie,
Goethe-Universität Frankfurt a. M.,
Max-von-Laue-Str. 9, 60438 Frankfurt



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.