














- DAZ.online
- DAZ / AZ
- DAZ 49/2011
- Neues zu Paracetamol - ...
Im Fokus
Neues zu Paracetamol - Hepatotoxischer Metabolit wirkt analgetisch
Mit großem medialen Interesse und Superlativen wurde in den vergangenen Tagen eine Publikation zum analgetischen Wirkmechanismus von Paracetamol [1] aufgenommen. Die Studie zeigt am Mausmodell, dass eine Aktivierung des Ionenkanals TRPA1 auf Rückenmarksebene in die analgetische Wirkung von Paracetamol involviert sein könnte.
TRPA1: der Schlüssel zum Verständnis?
TRPA1 ist ein nichtselektiver Ionenkanal aus der inzwischen 7 Subfamilien umfassenden Familie der TRP-Kanäle (transient receptor potential channels) und ist der bisher einzige identifizierte Vertreter der TRPA-Subfamilie. Bekannt war bisher, dass TRPA1 vor allem auf Nozizeptoren sensorischer Neurone wie C- und A∂-Fasern exprimiert ist und durch eine Reihe proalgetischer Stimuli wie Bradykinin, Acrolein, Senföl, Wasserstoffperoxid und Acetaldehyd aktiviert wird. Dementsprechend sah man eine wesentliche Funktion von TRPA1 vor allem auf Ebene der Depolarisation von Nozizeptoren und der Aus lösung eines Entzündungsschmerzes [2, 3]. Während einige Agonisten entsprechend dem klassischen Schlüssel-Schloss-Prinzip mit TRPA1 interagieren, bewirken andere elektrophile Substanzen eine kovalente Modifikation des Ionenkanals an nukleophilen Cysteinresten [4]. Die Aktivierung wird in diesen Fällen nicht von der Struktur sondern von der Re aktivität bestimmt.
Andersson et al. adressierten in ihrer Arbeit gezielt die Rolle von TRPA1 bei der Schmerz weiterleitung auf Rückenmarks ebene und kamen zu folgendem überraschenden Ergebnis:
Paracetamol wirkt bei der Maus nach systemischer (intraperitonealer) oder lokaler (intrathekaler) Gabe erwartungsgemäß analgetisch. Werden jedoch TRPA1-defiziente Mäuse (TRPA1 -/-) verwendet, bleibt der antinozizeptive Effekt von Paracetamol aus. Als Modell wurde der Hot-Plate-Test gewählt, bei dem die zeitliche Latenz gemessen wird, mit der Mäuse ihre Pfoten von einer 53 ºC warmen Metallplatte zurückziehen. Ein Ausbleiben dieser Reaktion nach 30 Sekunden wird als analgetische Wirkung gewertet. Im Falle der systemischen Paracetamol-Gabe konnten diese Befunde in einem weiteren Schmerzmodell (Writhing-Test) verifiziert werden.
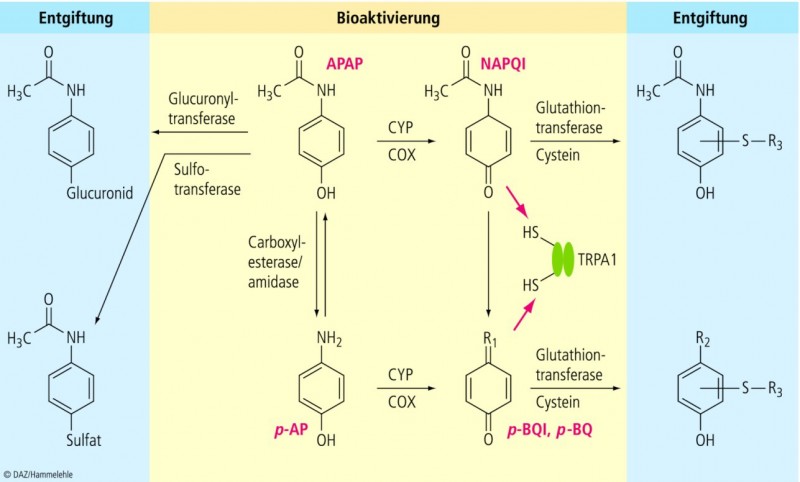
Paracetamol selbst zeigt in vitro keine stimulatorische Wirkung auf TRPA1, wohl aber seine elektrophilen Metabolite N-Acetyl-p-Benzochinonimin (NAPQI) und p-Benzochinon (Abb. 1). Die hierbei für NAPQI registrierten EC50 -Werte lagen im hohen nano- bis niedrigen mikromolaren Konzentrationsbereich.
NAPQI und p-Benzochinon wirken selbst analgetisch im Hot-Plate-Test, zeigen wie Paracetamol aber keine Wirkung bei TRPA1-defizienten Mäusen.
Nach Applikation von Paracetamol lässt sich ein stabiles Produkt von NAPQI (L-Cysteinyl-Konjugat) im Rückenmark nachweisen.
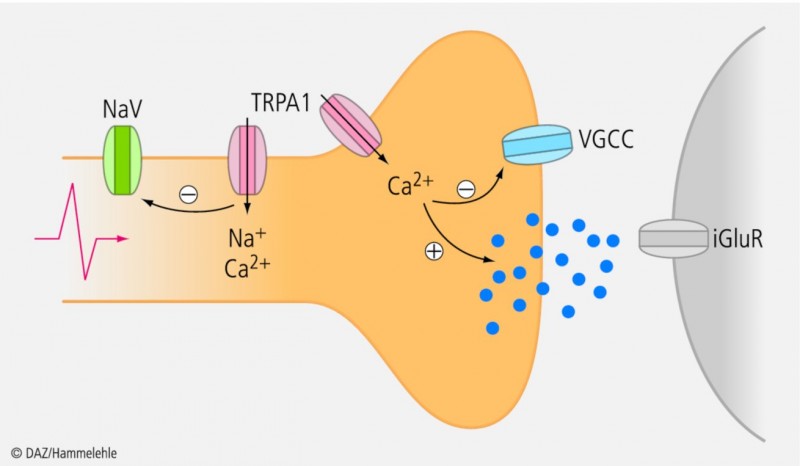
Erste elektrophysiologische In-vitro-Befunde sprechen dafür, dass die TRPA1-Aktivierung durch p-Benzochinon mit einer verminderten Erregbarkeit (Hemmung spannungsabhängiger Natrium- und Calciumkanäle) von Hinterhornneuronen einhergeht. Die Folge könnte eine verminderte Neurotransmitterfreisetzung – hier vor allem des für die Schmerzweiterleitung wichtigen Glutamats – sein (Abb. 2).
Kommentare und Fazit:
Die von Andersson et al. publizierte Arbeit stellt eine Anreihung gut konzeptionierter, aussagekräftiger Experimente dar. Zweifelsfrei konnte erstmals gezeigt werden, dass eine Aktivierung von TRPA1 auf spinaler Ebene in den gewählten tierexperimentellen Schmerzmodellen eine analgetische Wirkung hervorruft. Insofern liefert die Studie wichtigen Input für die Entwicklung neuer analgetischer Substanzen.
Die Wirkung in anderen experimentellen Systemen bleibt jedoch vorerst offen. Wie die Autoren selbst kritisch anmerken, zeigen die für solche Tests essenziellen TRPA1-Knock-out-Mäuse per se eine verminderte Sensitivität in verschiedenen Schmerzmodellen, die unter Einsatz mechanischer oder Kältereize arbeiten.
Trotz eines weiteren potenziellen Wirkmechanismus bleibt Paracetamol ein vergleichsweise schwaches Analgetikum mit geringer therapeutischer Breite. Gerade der letzte Punkt wird dankenswerterweise sehr offen diskutiert. So lassen die Autoren keinen Zweifel daran, dass eine Erhöhung der lokalen Bildung der analgetischen elektrophilen Metabolite durch Paracetamol-Dosissteigerung unweigerlich mit erhöhtem Gewebeschaden einhergeht. Als Perspektive wird vielmehr eine weiterführende Testung nicht elektrophiler TRPA1-Agonisten angestrebt. Inwieweit sich diese Substanzen, wie das in der vorliegenden Studie untersuchte nicht-psychoaktive Cannabinoid Δ9-Tetrahydro-cannabiorcol, als effiziente Analgetika etablieren können, bleibt abzuwarten.
Kritisch zu hinterfragen bleibt, warum die für NAPQI und p-Benzochinon in vivo ermittelte analgetische Potenz (NAPQI > p-Benzochinon) nicht der für die Metaboliten in vitro ermittelten Rangfolge bei der TRPA1-Aktivierung entspricht. Hinzu kommt, dass der Befund einer TRPA1-Aktivierung durch den Paracetamol-Metaboliten NAPQI keineswegs neu ist. Dieser Punkt wird in der Arbeit durch Andersson et al. eher am Rande erwähnt. So konnte in einer ein Jahr zuvor publizierten Studie [5] gezeigt werden, dass eine NAPQI-getriggerte TRPA1-Stimulation via Freisetzung proinflammatorischer Neuropeptide (Substanz P, Calcitonin Gene-Related Peptide) aus sensorischen Nervenendigungen zu einer neurogenen Entzündung der Atemwege bei der Ratte führt. Auch hier ließen sich nach Gabe therapeutischer Paracetamol-Dosen Spiegel von NAPQI in der Lunge nachweisen. Die Autoren diskutieren diese Befunde als möglichen Risikofaktor bei der Entstehung von Asthma. Zur Erinnerung: Epidemiologische Studien weisen auf ein erhöhtes Asthmarisiko nach Gabe von Paracetamol während Schwangerschaft, Kindheit, Jugend und bei Erwachsenen hin [6]. Ein kausaler Zusammenhang konnte bisher noch nicht schlüssig gezeigt werden.
Der von Anderson et al. präsentierte Mechanismus reiht sich in eine Reihe weiterer in den letzten Jahren publizierter Arbeiten ein. Erinnert sei an die noch stärkere Euphorie im Zusammenhang mit der vor ca. einem Jahrzehnt identifizierten "Cyclooxygenase-3" als spezifisches Target von Paracetamol [7] – die Hypothese wurde aus verschiedenen plausiblen Gründen [8] schnell fallen gelassen. Aber auch Untersuchungen zur möglichen Involvierung des Endocannabinoid-Systems haben das Stadium der tierexperimentellen Forschung bisher nicht überschreiten können. Der Theorie zufolge soll hier der Paracetamol-Metabolit p-Aminophenol (Abb. 1) eine Zwischenstufe bei der Bildung eines Fettsäureamidohydrolase-Hemmstoffs (AM-404) darstellen, der via Akkumulation des Endocannabinoids Anandamid zur analgetischen Wirkung von Paracetamol beitragen könnte [9]. Ein weiterer potenziell analgetischer Paracetamol-Effekt, nämlich die Aktivierung des absteigenden antinozizeptiven serotoninergen Systems und die sich daraus ergebende Interaktion zwischen Paracetamol und den Setronen (5-HT3-Antagonisten) konnte wiederholt überzeugend in experimentellen humanen Schmerzmodellen [10,11] belegt werden, lässt sich offensichtlich aber nicht auf alle Schmerzzustände übertragen [12,13]. Nicht zuletzt sprechen bereits Ende der 1990er Jahre publizierte Befunde für einen wesentlichen Beitrag der Cyclooxygenase-Hemmung in die analgetische Wirkung von Paracetamol. So bewirkt Paracetamol eine Hemmung der durch einen peripheren Entzündungsreiz stimulierten Prostaglandin-Synthese im spinalen Hinterhorn der Ratte [14]. Eine durch Paracetamol ausgelöste analgetische Wirkung auf Rückenmarksebene ist somit keineswegs neu!
DEBATTE
Paracetamol: Diskussion um Sicherheit und VerschreibungspflichtAls Ende 2010 der Erlanger Pharmakologe Prof. Dr. Dr. Kay Brune in einem Gastkommentar in der DAZ aufgrund neuer Studien gefordert hatte, Paracetamol entweder ganz vom Markt zu nehmen oder der Verschreibungspflicht zu unterstellen, schlugen die Wellen hoch. Wir haben die Argumente für und gegen Änderungen des Zulassungsstatus von Paracetamol in mehreren Beiträgen in der DAZ veröffentlicht und ein Dossier für Ihre eigene Meinungsbildung zusammengestellt. |
Zusammenfassend erscheint eine multifaktorielle Analgesie durch Paracetamol prinzipiell nicht abwegig, bedarf jedoch weiterer Klärung. Diese Bemühungen ändern jedoch nichts an dem vergleichsweise schwachen analgetischen Effekt der Verbindung. Trifft der neu identifizierte Signalweg partiell zu, würde dies zugleich analgetische und heptatotoxische/gewebeschädigende Wirkung von Paracetamol in einen mechanistischen Zusammenhang bringen. Wie eng das therapeutische Fenster zwischen beiden Komponenten ist, macht eine aktuelle Studie zu schweren Leberschädigungen infolge wiederholter geringfügiger Überdosierung mit Paracetamol [15,16] einmal mehr deutlich. In jedem Falle erscheint es verfrüht, den analgetischen Wirkmechanismus von Paracetamol – wie auch sein Nebenwirkungsspektrum (!) – als "aufgeklärt" zu betrachten.
Literatur[1] Andersson DA et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ9 -tetrahydrocannabiorcol. Nat Commun 2011 Nov 22;2:551. doi: 10.1038/ncomms1559.[2] McNamara CR et al. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A 2007;104:13525 – 30.[3] Bautista DM et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006;124:1269 – 82. [4] Hinman A et al. TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A 2006;103:19564 – 8. [5] Nassini R et al. Acetaminophen, via its reactive metabolite N-acetyl-p-benzo-quinoneimine and transient receptor potential ankyrin-1 stimulation, causes neurogenic inflammation in the airways and other tissues in rodents. FASEB J 2010;24: 4904 – 16. [6] Etminan M et al. Acetaminophen use and the risk of asthma in children and adults: a systematic review and metaanalysis. Chest 2009;136:1316 – 23. [7] Chandrasekharan NV et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A 2002;99: 13926 – 31. [8] Kis B et al. Acetaminophen and the cyclooxygenase-3 puzzle: sorting out facts, fictions, and uncertainties. J Pharmacol Exp Ther 2005;315: 1 – 7. [9] Högestätt ED et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem 2005;280:31405 – 12.[10] Pickering G et al. Analgesic effect of acetaminophen in humans: first evidence of a central serotonergic mechanism. Clin Pharmacol Ther 2006;79:371– 8. [11] Bandschapp O et al. Tropisetron blocks analgesic action of acetaminophen: a human pain model study. Pain 2011;152:1304 – 10. [12] Jokela R et al. The influence of ondansetron on the analgesic effect of acetaminophen after laparoscopic hysterectomy. Clin Pharmacol Ther 2010;87:672 – 8. [13] Minville V et al. Ondansetron does not block paracetamol-induced analgesia in a mouse model of fracture pain. Br J Anaesth 2011;106:112-8. [14] Muth-Selbach US et al. Acetaminophen inhibits spinal prostaglandin E2 release after peripheral noxious stimulation. Anesthesiology 1999;91:231– 9.[15] Craig DG et al. Staggered overdose pattern and delay to hospital presentation are associated with adverse outcomes following paracetamol-induced hepatotoxicity. Br J Clin Pharmacol 2011 Nov 22. [Epub ahead of print] [16] Akutes Leberversagen: Hohes Risiko bei wiederholter Paracetamol-Überdosierung. DAZ 2011; Nr. 47, S. 56 – 57
AutorProf. Dr. rer. nat. Burkhard HinzDirektor, Institut für Toxikologie und PharmakologieUniversitätsmedizin RostockSchillingallee 70, 18057 Rostock



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.