














- DAZ.online
- DAZ / AZ
- DAZ 49/2011
- Mit securPharm gegen ...
Arzneimittelsicherheit
Mit securPharm gegen Arzneimittelfälschungen
Im weltweiten Handel sind Arzneimittelfälschungen ein riesiges Problem. Die Zollstatistik der EU gibt für 2010 etwa 1800 Beschlagnahmungsfälle an den EU-Außengrenzen an. Dabei wurden etwa 3,2 Millionen gefälschte Arzneimittel im Originalwert von 26,6 Millionen Euro sichergestellt. Es bleibt offen, wie viele Fälschungen nicht gefunden wurden. Im Vergleich zum weltweiten illegalen Handel erscheinen die Fälschungen in der legalen Vertriebskette minimal. Zwischen 1996 und 2008 wurden nach Angaben des Bundeskriminalamtes 40 Fälle mit Bezug zum legalen deutschen Arzneimittelvertrieb bekannt. Dabei ging es meist um Fälschungen der Verpackungen. Doch die große Anzahl der Fälschungen im illegalen Handel stellt eine zunehmende Belastung für die Sicherheitsmechanismen des legalen Marktes dar. Es erscheint daher folgerichtig, die legale Vertriebskette noch wirksamer abzuschotten, um diese auch in Zukunft sicher zu erhalten.
Richtlinie 2011/62/EU
Dies dürfte eine wichtige Motivation für die neuen Regelungen der EU gegen Arzneimittelfälschungen sein. Die Richtlinie 2011/62/EU des Europäischen Parlamentes und des Rates vom 8. Juni 2011 wurde am 1. Juli 2011 veröffentlicht. Sie ist Teil des Ende 2008 konzipierten EU-Pharmapaketes zur Arzneimittelsicherheit. Ziel der EU-Richtlinie ist, die Patienten noch besser vor gefälschten Arzneimitteln zu schützen. Als Zusatznutzen sollen die neuen Maßnahmen Rückrufe unterstützen und die Warenbewirtschaftung durch maschinenlesbare Produktdaten erleichtern. Der letztgenannte Aspekt mag als Ausgleich für die zusätzlichen Kosten der Beteiligten verstanden werden. Die EU-Richtlinie enthält zwei zentrale Forderungen. Sie fordert
erstens Sicherheitsmerkmale, die es Großhändlern und Apotheken ermöglichen, die Echtheit des Arzneimittels zu überprüfen und einzelne Packungen zu identifizieren, sowie
zweitens eine "Vorrichtung, die es ermöglicht, zu überprüfen, ob die äußere Umhüllung manipuliert worden ist".
Die erste Forderung läuft auf ein individuelles Erkennungsmerkmal ("unique identifier") mit einer Nummer für jede einzelne Arzneimittelpackung heraus. Die zweite Bedingung erfordert ein Anti-Manipulationsmerkmal ("tamper proof evidence"), über das der Hersteller entscheiden soll. Dafür bieten sich Klebesiegel oder -punkte, Hüllen oder per forierte Öffnungslaschen an.
Delegierter Rechtsakt
Die Umsetzung wird durch einen delegierten Rechtsakt konkretisiert. Dieser ist eine ergänzende Regelung ohne Gesetzescharakter, die die EU-Kommission aufgrund einer Befugnis in der EU-Richtlinie erlassen darf. Die Veröffentlichung eines Konzeptpapiers am 18. November 2011 durch die EU-Kommission kann als offizieller Start für die Debatte über die Inhalte des de legierten Rechtsaktes betrachtet werden, dessen Verabschiedung Anfang 2014 erwartet wird.
Als einzig mögliches Erkennungsmerkmal betrachtet die EU-Kommission eine "Seriennummer", die für jede einzelne Packung nach dem Zufallsprinzip erzeugt wird. Damit lässt sich jede Packung eindeutig identifizieren – sie wird zu einem Unikat. Gemäß dem jüngsten Konzeptpapier könnten zusätzlich das Verfalldatum und eine nationale Nummer zur Produktkennzeichnung wie die deutsche Pharmazentralnummer angegeben werden. Maschinenlesbare Verfalldaten und Chargenbezeichnungen würden die Warenbewirtschaftung in der Apotheke erleichtern. Diese Daten können nach den Vorstellungen der EU-Kommission mit drei alternativen Verfahren maschinenlesbar auf die Packung aufgebracht werden: mit eindimensionalen Barcodes, mit zweidimensionalen Codes wie auf elektronischen Briefmarken oder als aufwendigste Variante mit der Radio-Frequenz-Identifikation (RFID).
In jedem Fall müssen die Seriennummern der einzelnen Packungen vom Hersteller in ein zentrales System eingegeben und bei der Abgabe in der Apotheke überprüft und ausgelesen werden. Als mögliche Varianten sieht die EU-Kommission zusätzlich stichprobenweise oder sogar verpflichtende Überprüfungen auf den dazwischen liegenden Handelsstufen, also insbesondere beim Großhandel, vor. Ein weiterer Aspekt, der gemäß dem Konzeptpapier der EU-Kommission im delegierten Rechtsakt geklärt werden muss, ist die Organisation der Datenbank, die die Seriennummern enthält. Möglich ist demnach eine Datenbank der beteiligten Akteure der Vertriebskette. Dabei müsste sichergestellt werden, dass die Daten über die Abgabe des Arzneimittels nicht für den Hersteller zugänglich sind. Als weitere Alternativen werden nationale Systeme unter staatlicher Verantwortung oder eine zentrale EU-weite Datenbank genannt. Bei Umsetzungen auf nationaler Ebene müssten Lösungen gefunden werden, um auch den Handel zwischen EU-Staaten zu erfassen.
Anwendungsbereich
Außerdem soll im delegierten Rechtsakt der verbindliche Anwendungsbereich des neuen Sicherheitssystems festgelegt werden. Dabei gelten gemäß der Richtlinie 2001/62/EU zwei Grundregeln:
Alle verschreibungspflichtigen Arzneimittel müssen die neuen Sicherheitsmerkmale tragen, sofern nicht festgestellt wird, dass für sie kein Fälschungsrisiko besteht. Dann werden sie auf einer "white list" verzeichnet.
Arzneimittel zur Selbstmedikation müssen die neuen Sicherheitsmerkmale nicht tragen, sofern nicht festgestellt wird, dass für sie ein Fälschungsrisiko besteht. Dann werden sie auf einer "black list" verzeichnet.
Kriterien für die Bewertung des Fälschungsrisikos sind der Preis, das Absatzvolumen und weitere spezifische Merkmale des Arzneimittels, die bisherigen Fälschungen des Produktes, der Schweregrad der Indikation sowie mögliche Gefahren für die öffentliche Gesundheit. Die daraufhin erstellten Ausnahmelisten ("black" und "white list") sollen mit dem delegierten Rechtsakt festgelegt werden. Sie sollen für die ganze EU einheitlich gelten. Eine freiwillige Anwendung durch den Hersteller soll gemäß dem Konzeptpapier vom 18. November 2011 nicht zulässig sein. Als Optionen für die Formulierung der Ausnahmelisten werden im Konzeptpapier Indikationen, Handelsnamen, Wirkstoffe oder flexible Varianten genannt. Als mögliche Lösung wird dort ein Punktesystem angedacht, das die Kriterien verknüpft.
securPharm-Initiative in Deutschland
Angesichts dieser Vorgaben sehen die Beteiligten keinen Raum mehr für grundlegende Diskussionen, ob ein solches System überhaupt wünschenswert ist. Diese Entscheidung ist auf europäischer Ebene bereits gefallen. Doch umso mehr stehen die Betroffenen jetzt vor der Herausforderung, die Zeit bis zum Erlass des delegierten Rechtsaktes zu nutzen, um praxistaugliche Lösungen zu erarbeiten, damit diese von der EU festgeschrieben werden können. Kritiker sehen insbesondere die Gefahr, dass die EU ein zentrales System einführen könnte, bei dem Abgabedaten, die heute privatwirtschaftlich ausgewertet werden, behördlich verwaltet würden und damit den Systembeteiligten nicht mehr zur Verfügung stünden. Wer die Daten verwaltet, hätte aber auch die Verantwortung für das einwandfreie Funktionieren des Systems. Im Extremfall könnte mit Hilfe der RFID-Technik ein "track-and-trace"-System eingeführt werden, mit dem die Handelswege jeder Packung zu verfolgen wären. Um solchen Entwicklungen vorzubeugen und um überflüssige Kosten und Mühen zu vermeiden, haben sich die betroffenen Verbände in Deutschland frühzeitig verständigt, ein gemeinsames Konzept zu entwickeln, das die Anforderungen der EU erfüllt, aber die möglichen unerwünschten Effekte vermeidet.
Dazu haben die ABDA, die Verbände der pharmazeutischen Industrie (BAH, BPI, Pro Generika und vfa) und der Großhandelsverband PHAGRO bereits im April 2011 die Gründung der gemeinsamen, unabhängigen und nicht gewinnorientierten Initiative securPharm beschlossen. Diese breit angelegte Partnerschaft ist auf gemeinschaftliche Entscheidungen aller Beteiligten ausgelegt. Die Vereinsgründung ist für den 16. Dezember 2011 geplant. Mitinitiator Dr. Reinhard Hoferichter erklärte zu den Aufgaben der Initiative: "Es geht darum, die Anforderungen des Gesetzgebers in der Praxis umzusetzen." Die Initiative securPharm habe sich zum Ziel gesetzt, für Deutschland ein System zur Umsetzung der EU-Richtlinie zu erarbeiten und 2013 in einem Pilotversuch zu testen.
Die Zusammenarbeit innerhalb der securPharm baut auf Konsens und gemeinsamen Zielen auf. Gegenüber der DAZ bezeichnete Hoferichter die securPharm als "Schiedsrichter und Regelüberwacher". Im Unterschied zu früheren Plänen habe man sich kürzlich auf ein dreiteiliges Konzept geeinigt. Dabei erstellen Apotheken und Hersteller eigenständig ihre jeweiligen Systeme und finanzieren diese auch selbst, während securPharm als Bindeglied für die ordnungsgemäße Verifizierung sorgt. So sei sichergestellt, dass jeder der Herr seiner Daten bleibt.
DataMatrix-Code
Ein zentrales Element des securPharm-Konzepts ist die Kodierung der Arzneimittelpackungen mit ei nem zweidimensionalen DataMatrix-Code (Abb. 1).
Foto: IFA
Der geplante Code enthält eine Produktnummer, die Chargenbezeichnung, das Verfalldatum und die Nummer der Packung. Für die Darstellung wurde der zweidimensionale Code ECC 200 gewählt, der aufgrund seiner Robustheit auch bei 30%iger Beschädigung lesbar ist. Die Seriennummer wird nur im Code enthalten sein, die übrigen Inhalte des Codes werden wie bisher auch in Klarschrift aufgedruckt. Die Produktnummer wird aus der nationalen Pharmazentralnummer (PZN) erzeugt. Durch einen Zusatz entsteht daraus eine international eindeutige "Pharmacy-Product-Number" (PPN). So können die Arzneimittel international eindeutig gekennzeichnet werden, ohne die national übliche PZN aufgeben zu müssen.
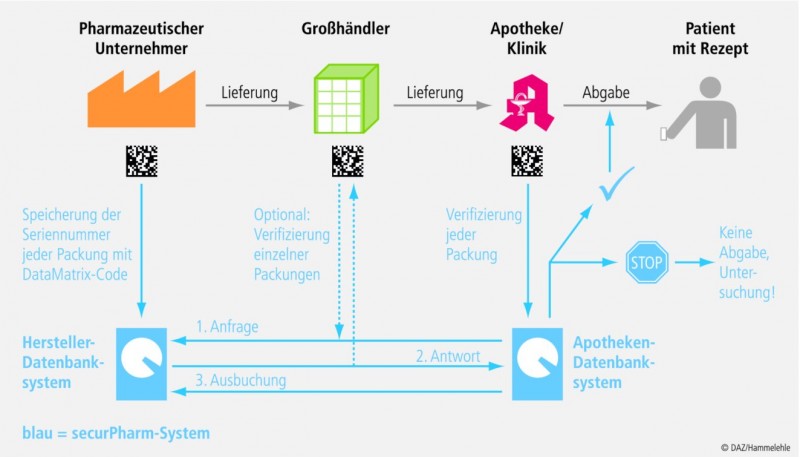
Funktionsweise des Systems
Zum System der Arzneimittelverifizierung (siehe Abb. 2) gehören drei Elemente:
- die eindeutige Kennzeichnung mit dem oben beschriebenen DataMatrix-Code,
- die Speicherung dieser Kennzeichnung in einer Datenbank und
- die Überprüfung der Packung durch die ab gebende Apotheke.
Wenn der Hersteller eine Packung in den Handel bringt, stellt er den zugehörigen Code in die Datenbank der Hersteller ein. Dann können die autorisierten Marktpartner auf diese Daten zugreifen, um die Authentizität der Packung zu prüfen.
Der Großhandel soll die Packung fakultativ prüfen können, beispielsweise in Verdachtsfällen. Entscheidend für die Funktionsweise des Systems ist aber der Zugriff der Apotheke auf die Datenbank beim Abgabevorgang, die "End-to-End"-Kontrolle. Dabei erhält die Apotheke die Bestätigung, dass die Packung im System bekannt ist. Daraufhin wird die Packung in der Datenbank als "abgegeben" vermerkt. Diese Statusänderung ist ein grundlegender Schritt für das Funktionieren des Systems. Denn bei späteren Abfragen mit derselben Packung gibt die Datenbank ein Warnsignal. Das ganze Verfahren funktioniert demnach nur, wenn jede Apotheke verpflichtet wird, jede Packung bei der Abgabe zu erfassen, weil sonst nicht sicher wäre, ob die Packung bereits irgendwo abgegeben wurde. Wenn ein Fälscher den Code einer Originalpackung mehrfach kopiert, würden die zweite und jede weitere Packung mit demselben Code eine Warnmeldung auslösen. In gleicher Weise würden unbekannte Packungen mit falschen Codes einen Alarm auslösen. Dann kann der Fälschungsverdacht geprüft werden.
Quelle: securPharm
Zweite Phase 2012
Die Kodierelemente und das Modell für die Arzneimittelverifizierung wurden bereits entwickelt. Anfang 2012 soll das Projekt in seine zweite Phase eintreten. Dann will securPharm der Industrie die Codespezifikation zur Verfügung stellen, damit die Packungen entsprechend gekennzeichnet werden können. Im Jahr 2012 sollen auch das Verifizierungsmodell in ein Pflichtenheft umgewandelt und die Teilnehmer für den Pilotversuch gewonnen werden. Dabei wird eine begrenzte Anzahl von Herstellern, Präparaten, Großhändlern und Apotheken angestrebt, die freiwillig teilnehmen und auf Modellregionen konzentriert sein sollen.
Pilotversuch 2013
Im Januar 2013 will securPharm den Pilotversuch beginnen und damit in die dritte Phase des Projektes eintreten. Der Pilotversuch soll etwa drei Mona te dauern und in den folgenden drei Monaten ausgewertet werden. Mit dem Pilotversuch will securPharm die technische Umsetzung des Systems prüfen und die Konzepte für die Kodierung und Verifizierung unter den Bedingungen des deutschen Marktes erproben. Dazu gehört auch die Kompatibilität mit der in Deutschland angewendeten Software. Letztlich soll die Praxistauglichkeit des Konzeptes unter Beweis gestellt werden, um sich gegenüber der Politik als Referenz für die For mulierung des delegierten Rechtsaktes zu präsentie ren. Nach Angaben von securPharm will Deutschland mit dem geplanten Pilotversuch das erste große EU-Land sein, das die Umsetzung testet.
Umsetzung ab 2017
Im Anschluss an den Pilotversuch soll sich securPharm vom Versuchskoordinator zur Betreibergesellschaft wandeln. Die Richtlinie 2011/62/EU gewährt zwar nur eine Umsetzungsfrist von 18 Monaten, aber die Sicherheitsmerkmale als Voraussetzung für das System sollen erst drei Jahre nach Veröffentlichung des delegierten Rechtsaktes vorgeschrieben werden. Die flächendeckende Umsetzung wird daher voraussichtlich Anfang 2017 verpflichtend sein.
Die Phase zwischen dem Pilotversuch und der flächendeckenden Umsetzung bezeichnete Hoferichter gegenüber der DAZ als besonders spannend. Das System könnte dann abgeschaltet und später mit einem "big bang" deutschlandweit neu starten. Hoferichter sieht allerdings Vorteile für eine andere Variante. Dabei würden die Versuchspartner auch nach dem offiziellen Ende des Pilotversuchs mit dem System weiterarbeiten. Dann könnten nach und nach weitere Teilnehmer hinzukommen, bis alle verpflichtend dabei sein müssen. Bei dieser Variante würde das System nach dem Pilotversuch nicht mehr abgeschaltet, sondern Detailänderun gen, die aufgrund der Versuchserfahrungen nötig werden, würden im laufenden Betrieb vorgenommen. So könnte das System schrittweise für den Betrieb im großen Maßstab fit gemacht werden.
Zeitplan für die vorgesehenen Maßnahmen zur Verbesserung der Fälschungssicherheit | |
2012 |
securPharm: Vorbereitung des Pilotversuchs, Weiterentwicklung der Kodierung und Verifizierung |
1. Halbjahr 2013 |
securPharm: Durchführung und Aus wertung des Pilotversuchs |
Anfang 2014 |
EU: Erlass des delegierten Rechtsaktes |
2013 bis 2016 |
securPharm: Vorbereitung der flächendeckenden Einführung |
2017 |
Flächendeckende Umsetzung der Arzneimittelverifizierung in Deutschland |
2017 |
3 Jahre nach Veröffentlichung des delegierten Rechtsaktes: verpflichtende Anwendung der Sicherheitsmerkmale |
Kosten des Projekts
Die EU-Richtlinie enthält keine genauen Angaben zur Verteilung der Kosten des Projekts, aber sie bestimmt, dass die Arzneimittelhersteller die Kosten für das Datenspeicher und -abrufsystem tragen müssen. Allein für die Hersteller verschreibungspflichtiger Arzneimittel rechnet die EU-Kommission mit einmaligen Anlaufkosten von 8,85 Mrd. Euro für alle Hersteller in der EU zusammen, davon 1,8 Mrd. Euro für Verpackungsmaschinen mit packungsindividueller Kennzeichnung. Außerdem werden EU-weit laufende Kosten von über 1 Mrd. Euro pro Jahr für die Hersteller erwartet.
Folgen für die Apotheken
Auch wenn die Verteilung der Kosten innerhalb der Industrie noch nicht geklärt ist, wird die In dustrie den weitaus größten Teil der Kosten tragen. Die Kosten für die Apotheken werden wesentlich davon abhängen, ob dort bereits moderne Scanner vorhanden sind, die zweidimensionale Codes lesen können. Bis 2017 dürfte dies in vielen Apotheken ohnehin gewährleistet sein. Hinzu kommen die Kosten für die Software in den Apotheken und der Kostenanteil der ABDA für die Betreiber gesellschaft.
Für den Alltag wichtiger dürften die Folgen für die Arbeitsabläufe sein. Bereits bei der Diskussion zu diesem Thema beim Deutschen Apothekertag 2011 in Düsseldorf wurde betont, dass die Abfrage in der Datenbank deutlich schneller sein muss als bei den Tests zum elektronischen Rezept. Dr. Peter Hohmann, Vorsitzender des Hessischen Apotheker verbandes, erklärte dort, die Abfrage solle in weniger als einer Sekunde möglich sein. Das teilweise langsame Internet in ländlichen Regionen könnte damit zu einem Hindernis werden.
Anwendung im Apothekenalltag
Aus Apothekenperspektive drängen sich weitere Überlegungen auf: Gegenüber den Apothekenkunden dürfte es eindrucksvoll sein und als positives Transparenzsignal wirken, wenn diese sich bei der Abgabe selbst von der positiven Antwort des Systems überzeugen können, vielleicht durch eine grüne Lampe. Apotheken, die die Datenbankabfrage überzeugend inszenieren, könnten sich so möglicher weise auch gegenüber Versandapotheken profilieren, die die Überprüfung nicht so präsentieren können. Andererseits wirft die Ausbuchung des Arzneimittels aus der zentralen Datenbank in Verbindung mit dem Abgabevorgang Probleme auf, beispielsweise wenn ein Patient erst im letzten Moment zu verstehen gibt, dass das Arzneimittel falsch gewählt oder unerwünscht ist. Dies spricht dafür, die Ausbuchung aus der Datenbank ganz am Ende des Kundengespräches vorzunehmen. Dann würde sie aber ein zu sätzlicher Schritt, der den Arbeitsablauf verlangsamt.
Dies ist nur einer von vielen Aspekten der praktischen Umsetzung, die im Pilotversuch geklärt werden müssen. So ist auch noch offen, was geschehen soll, wenn das System eine unbekannte Packung meldet. Wie soll sich das Apothekenteam bei einem Fälschungsverdacht verhalten? Große praktische Bedeutung für die Apotheken dürfte der Umgang mit versiegelten Packungen haben. Die Apotheken müssen Fertigarzneimittel stichprobenweise prüfen, Apotheker sollten zudem die Packungsbeilage lesen können und sich einen Eindruck vom Aussehen der Arzneiform oder von Applikationshilfen machen können. Denn nur so ist eine patientenbezogene Beratung vielfach erst möglich. Dies erfordert eine Möglichkeit zur nachträglichen Versiegelung, die aber auch vertrauenswürdig sein muss.
Dieses Problem betrifft auch Parallelimporteure, die ein Siegel zwangsläufig zerstören müssen. Dazu fordert die EU-Richtlinie, das zerstörte Siegel durch ein "gleichwertiges" Sicherheitsmerkmal zu ersetzen. Was "gleichwertig" bedeutet, soll im delegierten Rechtsakt festgelegt werden. Außerdem müssen Umverpacker die Originalseriennummer in der Datenbank löschen und eine neue Nummer für jede Packung vergeben. Eine weitere Frage ist, wie das auf Packungen ausgelegte Konzept bei der Zweitverblisterung umgesetzt werden kann.
Offene Fragen
Weitere Fragen betreffen die white list mit den Ausnahmen von der neuen Regelung. Für sehr billige Generika mit Herstellerabgabepreisen im Centbereich dürfte kaum ein Fälschungsrisiko bestehen, andererseits wären die Kosten für das Aufbringen und Verwalten des Codes hier unverhältnismäßig hoch. Die Position der betroffenen Hersteller könnte dabei gespalten sein, denn Ausnahmen für nieder preisige Produkte könnten deren Image schaden, wenn die Sicherheitsabfrage in der Apotheke für die Patienten sichtbar ist. Ausnahmen für niederpreisige Packungen werden auch im Konzeptpapier der EU-Kommission vom 18. November 2011 thematisiert. Dort wird ein Herstellerabgabepreis von über 2 Euro als mögliche Grenze für einen "hohen Preis" angedacht. Die Unterschiede zwischen fälschungsanfälligen Originalprodukten und eher wenig fälschungsgefährdeten Generika werfen auch Fragen zur Kostenverteilung auf. Packungsbezoge ne Verrechnungen gingen dann zulasten der Generika, soweit sie nicht auf der white list stehen.
Auf weitere mögliche Probleme hat auch die EU-Kommission im Konzeptpapier vom 18. November 2011 hingewiesen. Dort wird ausdrücklich der Schutz von persönlichen Daten und Geschäftsgeheimnissen erwähnt. Neben Patientendaten müssten auch Daten über die Anzahl hergestellter Packungen, den Ort der Arzneimittelabgabe und den Ort des Umpackens geschützt werden. So bleiben bei der Betrachtung von Details noch viele Fragen offen – für das System insgesamt und für die Umsetzung in der Apotheke. Die DAZ wird die Entwicklung daher weiter verfolgen.
Wünsche eines Apothekers an die EU-Kommission und an securPharm
Die Apotheken sind durch das AMNOG wirtschaftlich stark belastet. Die Arbeitsabläufe bei der Arzneimittelabgabe leiden dauerhaft unter der Umsetzung der Rabattverträge. Die neue Apothekenbetriebsordnung wird voraussichtlich zu weiteren Anforderungen in der Beratung und an anderen Stellen des Apothekenbetriebs führen. Der Spielraum für zusätzliche technische und bürokratische Anforderungen und die Akzeptanz für neue Aufgaben sind daher sehr begrenzt. Die Arzneimittelsicherheit und damit auch die Fälschungssicherheit sind wichtige Ziele, denen sich die Apotheker verpflichtet fühlen. Doch die Umsetzung des geplanten Modells zur Authentifizierung von Packungen kann nur gelingen, wenn dies praxisnah und alltagstauglich gestaltet ist. Darum richtet ein Apotheker aus der Praxis folgende Wünsche an securPharm:
1. Lasst mich Apotheker bleiben! Ich möchte weiterhin die Verpflichtung zur stichprobenweisen Prüfung von Fertigarzneimitteln erfüllen (können). Ich möchte Packungsbeilagen lesen, Applikationshilfen ansehen und die Form und Farbe von Dragees und Tabletten selbst sehen können. Ich möchte nicht, dass der böse Spruch vom Schachtelverkäufer wahr wird, sondern ich möchte auch in Zukunft wissen, was in den Schachteln steckt.
2. Schafft keine neuen Probleme für das Beratungsgespräch! Wenn alle Folgen der Rabattverträge mit dem Patienten besprochen sind, ist oft kaum noch Zeit für die pharmazeutische Beratung. Darum muss die Authentifizierung der Packung schnell und einfach sein. Sie darf keine neuen Arbeitsabläufe und keine neuen Probleme schaffen. Dazu sollte jede Packung nur einmal gescannt werden müssen. Damit sollte die Packung aber noch nicht als "abgegeben" vermerkt werden. Dies sollte erst nach einer Bestätigung ganz am Ende des Beratungsgespräches geschehen. Vielleicht war es ja doch die falsche Packung, oder der Kunde überlegt es sich im letzten Moment anders.
3. Lasst die Technik nicht die Apotheke beherrschen! Auch die beste EDV funktioniert nicht immer fehlerfrei. Es gibt Stromausfälle und andere unerwartete Probleme. Außerdem machen auch Menschen manchmal Fehler. Darum braucht auch dieses System eine praktikable Möglichkeit zur Korrektur.
4. Übertreibt es nicht! Fälschungen sind weltweit ein großes Problem, aber wer fälscht Packungen, die nur ein paar Cent kosten? Der Aufwand sollte also nicht größer gemacht werden als nötig. Verhältnismäßigkeit ist eine grundsätzliche Forderung an jede Verwaltung.
5. Speichert nur die Daten, die wirklich nötig sind! Der Auftrag zur Authentifizierung der Packungen ist klar beschrieben. Es geht darum, die Packungen zu erkennen, aber es interessiert nicht, welchen Weg sie nehmen und wer sie abgibt. Die Apotheken-EDV ist eine gefährliche Schnittstelle. Hier könnte verknüpft werden, welcher Patient welche Packung erhält. Doch dies geht nur die Apotheker als Heilberufler an. Die Industrie oder der Großhandel dürfen dies nicht erfahren. Auch Rückrufe bis zum Patienten verfolgen zu wollen, ist nicht erstrebenswert. Ein System für die Arzneimittelsicherheit darf nicht das Vertrauensverhältnis zu den Patienten gefährden. Sonst würde es mehr schaden als nutzen. Darum darf nicht gespeichert werden, welche Apotheke eine bestimmte Packung abgegeben hat. Wenn die Datenstruktur keine Apothekendaten enthält, können diese auch nicht mit Patientendaten verknüpft werden.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.