


















- DAZ.online
- DAZ / AZ
- DAZ 1/2011
- Die Brücke zwischen ...
Molekularbiologie
Die Brücke zwischen Genom und Umwelt
Alte …
Der Begriff "Epigenetik" geht auf den englischen Entwicklungsbiologen Conrad Hal Waddington (1905 – 1975) zurück. Die griechische Vorsilbe epí bedeutet "darüber" oder "auf". Man kennt sie vielleicht von Begriffen wie "Epithel", "Epidermis" oder "Epidemiologie". Waddington bezeichnete als "Epigenese" Wachstum und Differenzierung eines Embryos, wobei die genetische Information mit anderen zellulären Bestandteilen der Zygote wechselwirkt [1]. Er war der Meinung, dass nicht bereits in der befruchteten Eizelle determiniert ist, wie der spätere Organismus aussehen wird, sondern dass noch zusätzliche Faktoren mit eine Rolle spielen, so dass sich der Phänotyp, also das Erscheinungsbild eines Organismus, aus der Summe von Genotyp und Epigenotyp zusammensetzt. Auf ihn geht die Gleichung zurück:
Epigenese + Genetik = Epigenetik
Wohlgemerkt: Diese Thesen entstanden um 1940, zu einem Zeitpunkt als man noch keine Ahnung von der Molekülstruktur der Nukleinsäuren hatte und zu einer Zeit, als George Beadle und Edward Tatum mit ihrer "Ein-Gen-ein-Enzym-Hypothese" wissenschaftlich erfolgreich waren. Mittlerweile kennt man die Struktur der DNA-Doppelhelix, und man weiß auch, dass die strenge Zuordnung eines Gens zu einem Enzym nicht stimmt, sondern dass ein Gen durchaus für mehrere, unterschiedliche Proteine codieren kann und dass es Gene gibt, die nur in RNA umgeschrieben werden.
… und neue Begriffserklärung
Heutzutage hat sich auch die Definition der Epigenetik etwas verändert hin zu "mitotisch und/oder meiotisch vererbbaren Änderungen in Genfunktionen, die nicht durch Änderungen der DNA-Sequenz verursacht sind, die aber wichtig für das Verständnis von Entwicklungsprozessen und phänotypischen Ausprägungen eines Organismus sind" [1].
Das heißt, dass nicht mehr nur die strikte Abfolge der Nukleinsäurebasen den Informationsgehalt des Genoms ausmacht, sondern dass es zusätzliche Modifikationen gibt, die entscheidend an der Ausbildung eines Organismus beteiligt sind. Damit lässt sich dann auch erklären, weshalb aus einer befruchteten Eizelle mit einer definierten genetischen Ausstattung während der nachfolgenden mitotischen Zellteilungen zur Ausbildung des menschlichen Organismus ca. 200 unterschiedliche Zelltypen entstehen können. Sehr einfach dargestellt kann man die Epigenetik mit einem Passwort-Schutz vergleichen: Obwohl in jeder Körperzelle dieselben Gene vorhanden sind, sind in einer bestimmten Zelle zu einem bestimmten Entwicklungsstadium nur ca. 10% der Gene aktiv, ganz ähnlich wie Datenbanken durch bestimmte Passwörter nur jeweils Teile der Information an verschiedene Nutzer freigeben.
Molekulare Grundlagen
Molekular stecken hinter dieser Epigenetik-Definition Methylierungen der DNA, posttranslationale Modifikationen an Histonproteinen sowie Chromatin-Umstrukturierungen. Vor allem DNA-Methylierungen und Histonmodifikationen sind bereits intensiv beforscht und haben auch schon zur Entwicklung von Arzneistoffen geführt.
Was heißt das? Dafür wollen wir uns noch einmal kurz die Struktur der Chromosomen vergegenwärtigen:
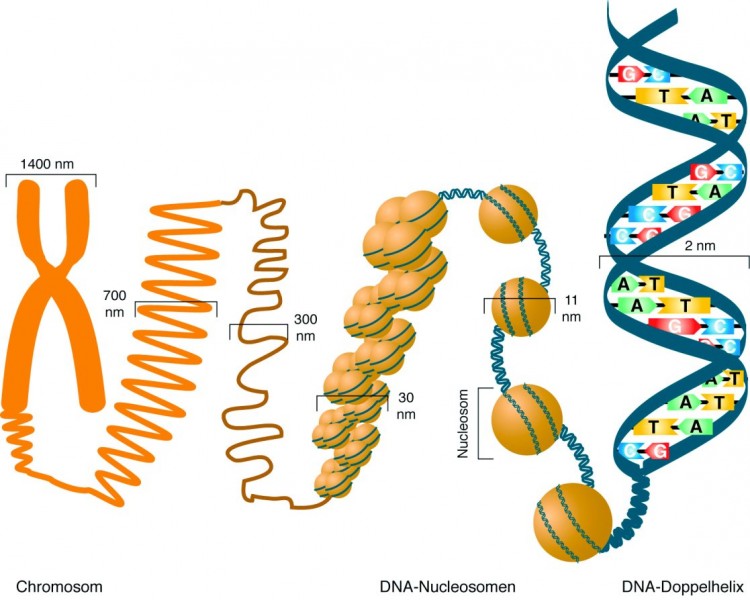
Jede menschliche Körperzelle enthält in ihren 46 Chromosomen 6 x 106 kbp DNA. Diese DNA hätte "ausgestreckt" eine Gesamtlänge von insgesamt knapp 2 m und muss deshalb sehr stark kompaktiert werden, um in den Zellkern zu passen (Abb. 1). Die wichtige erste Verpackungseinheit hierbei stellen die sogenannten Nucleosomen dar: Ein Oktamer aus je zwei Molekülen der Histonproteine H2A, H2B, H3 und H4, um das sich 147 bp DNA wickeln. Ein weiteres Histon, H1, wird als Linker-Histon bezeichnet und bindet an die DNA zwischen zwei Nucleosomen. Diese Verpackungseinheiten werden dann noch weiter aufspiralisiert und kompaktiert und bilden letztlich das dreidimensionale Gebilde des Chromatins im Zellkern.
Histone des Core-Oktamers sind basische Proteine, die zwischen den verschiedenen eukaryontischen Organismen extrem konserviert und aus relativ vielen Lysinen und Argininen aufgebaut sind. Die positiven Ladungen dieser Aminosäureseitenketten können sehr effektiv die negativen Ladungen des DNA-Zuckerphosphatrückgrats neutralisieren. Über zahlreiche Wasserstoffbrücken, aber auch durch hydrophobe Wechselwirkungen und Salzbrücken wird der DNA-Protein-Komplex zusammengehalten. Die Histone des Oktamers sind überwiegend globulär aufgebaut, verfügen jedoch jeweils über ein sehr flexibles N-terminales Ende, das aus dem Komplex herausragt und zugänglich für die verschiedenen posttranslationalen Modifikationen ist.
DNA-Methylierung …
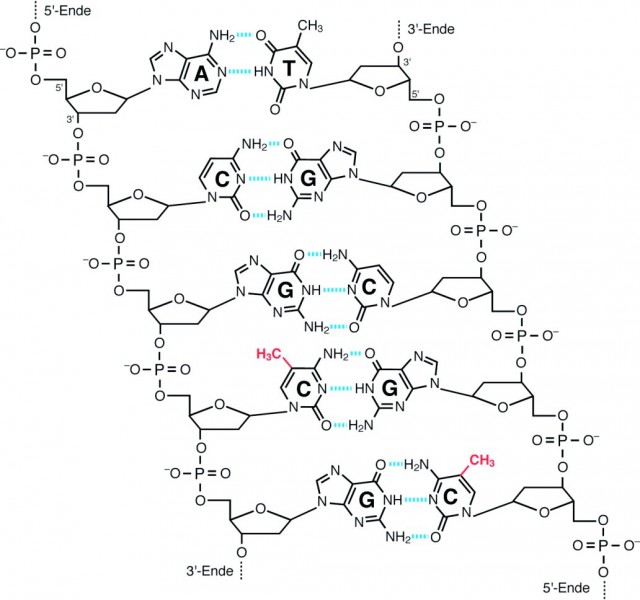
Bei der DNA-Methylierung wird die DNA-Base Cytosin vor allem innerhalb eines 5-Cytosin-Guanin-3-Dinukleotids (CpG-Dinukleotid) zu 5-Methylcytosin modifiziert (Abb. 2). Da der komplementäre Strang an der Stelle ebenfalls ein CpG-Dinukleotid aufweist, sind immer beide Stränge der Doppelhelix an der spezifischen Stelle methyliert. Die Methylierung von Cytosin ändert nichts an der Basenpaarung mit Guanin, so dass diese Modifikation das Codierungsverhalten der DNA nicht ändert. Allerdings verändert die Methylierung die Bindungsaffinität von DNA-bindenden Proteinen und beeinflusst dadurch die Genexpression – also die Realisierung der genetischen Information.
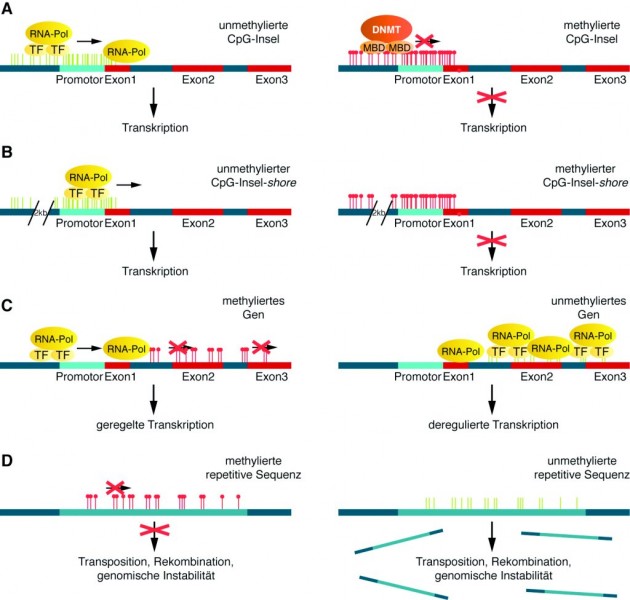
Über das gesamte menschliche Genom – in Gensequenzen ebenso wie in Transposons, also springenden Genbereichen, und in Sequenzbereichen zwischen den Genen – findet man DNA-Methylierungen [2]. Eine besondere Betrachtung verdienen hingegen die CpG-Inseln und die sogenannten "Shores" der CpG-Inseln.
CpG-Inseln sind dadurch charakterisiert, dass sie mehr als 200 Basenpaare lang sind und zu mindestens 50% aus C- und G-Nukleotiden bestehen. Ca. 60% der Promotoren menschlicher Gene sind mit derartigen CpG-Inseln assoziiert und ca. 6% dieser CpG-Inseln werden Zelltyp-spezifisch während der Embryonalentwicklung und Gewebedifferenzierung methyliert [3]. Ungefähr 2000 Basen entfernt von diesen CpG-Regionen hat man Bereiche identifiziert, die ebenfalls CpG-Dinukleotide, allerdings in geringerer Häufigkeit, enthalten. Diese Regionen werden alsCpG-Insel-Shores bezeichnet. Während sich die DNA-Methylierung in den CpG-Inseln des Promotorbereichs und auch in den CpG-Insel-Shores eher negativ auf die Transkription des nachgeschalteten Gens auswirkt, ist die Methylierung innerhalb der Genbereiche für eine korrekte Transkription wichtig (Abb. 3) [2, 3].
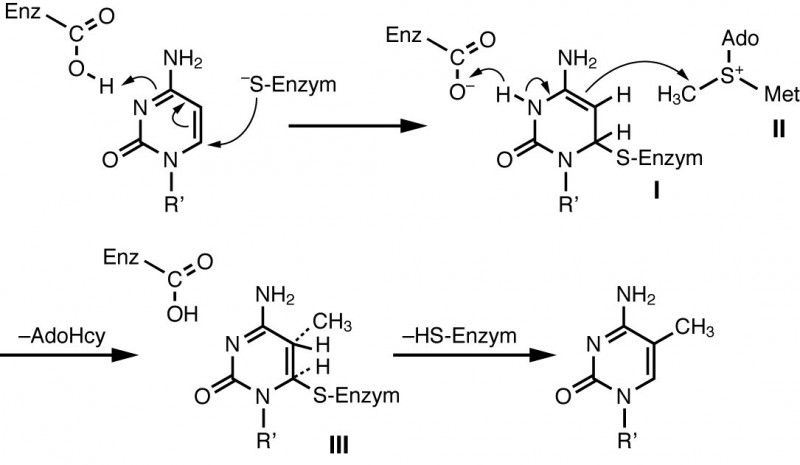
Im Menschen wurden vier Enzyme gefunden, die für die DNA-Methylierung verantwortlich sind: die DNA-Methyltransferasen DNMT1, DNMT2, DNMT3A und DNMT3B. Die DNMT2 spielt eher eine untergeordnete Rolle. Während DNMT3A und 3B DNA de novo methylieren, hat DNMT1 eine Präferenz für hemi-methylierte DNA (DNA, die nur auf einem Strang methyliert ist) und sorgt nach der semikonservativen Replikation der DNA für die Weitergabe der Methylierungsmuster. Dabei verknüpft sich das Enzym mit Cytosin zu einem Addukt, das über S-Adenosylmethionin (SAM) methyliert wird (Abb. 4). SAM entsteht aus der Aminosäure Methionin, deren Verfügbarkeit wiederum von Betain und Folsäure beeinflusst wird.
Neben der Methylierung in CpG-Dinukleotiden hat man mittlerweile im Menschen auch Methylierungen innerhalb von CHG- bzw. CHH-Sequenzen gefunden, wobei "H" die Basen Adenin, Cytosin oder Thymin repräsentiert. Außerdem kann man in einigen Zellen neben 5-Methyl-2‘-deoxycytidin auch 5-Hydroxymethyl-2‘-deoxycytidin finden. Die Funktionen dieser DNA-Modifikationen sind allerdings noch relativ wenig erforscht.
… und ihre Bedeutung im gesunden …
Erstaunlicherweise hat die DNA-Methylierung dramatische Effekte auf den Organismus:
Bereits kurz nach der Befruchtung wird über das "genomic imprinting" festgelegt, ob die väterliche oder die mütterliche Kopie eines geprägten Gens exprimiert wird (siehe Kasten "Genomic Imprinting").
Im wenige Tage alten weiblichen Embryo wird in jeder Zelle jeweils eines der beiden X-Chromosomen inaktiviert, wobei neben anderen Mechanismen auch die DNA-Methylierung eine Rolle spielt.
Ohne korrekte DNA-Methylierung, die an der kontrollierten Expression bestimmter Gene beteiligt ist, kann sich der menschliche Organismus nicht normal entwickeln und funktionieren. Das erklärt auch, weshalb sich das Methylierungsmuster in verschiedenen Geweben eines Organismus unterscheidet.
Methylierte repetitive Elemente (Transposons) sind inaktiv und immobil und beeinträchtigen dadurch nicht die Chromosomenstabilität.
Interessante Aspekte bringt die Untersuchung der DNA-Methylierung bei eineiigen Zwillingen. Schon lange hat man versucht, über den Vergleich von getrennt und zusammen aufgewachsenen eineiigen Zwillingen den Einfluss der Umwelt auf die Entwicklung festzustellen. Durch die Epigenomik hat sich hier noch eine andere Ebene eröffnet. Und tatsächlich konnte festgestellt werden, dass eineiige Zwillinge im Kleinkindalter eine sehr ähnliche DNA-Methylierung aufweisen, während sich das Methylierungsmuster bei älteren eineiigen Zwillingen deutlicher unterschied. Diese Unterschiede waren umso markanter, je kürzer die Zwillinge zusammengelebt hatten und je unterschiedlicher ihre Lebensweisen waren [4].
Im Rahmen des Human Epigenome Project sollen die DNA-Methylierungsmuster der Gene in den verschiedenen menschlichen Geweben identifiziert werden (www.epigenome.org).
… und im kranken Organismus
Ganz allmählich erkennt man immer mehr die Zusammenhänge zwischen der Epigenetik und Krankheiten. Generell sind hier jedoch unterschiedliche molekulare Mechanismen auseinanderzuhalten: Zum einen können Mutationen in Proteinen auftreten, die die methylierte DNA erkennen und dann bestimmte Effekte auslösen. Das heißt, hier sind zwar die Methylierungszeichen richtig gesetzt, aber die Moleküle, die diese Zeichen interpretieren müssen, können es nicht (mehr) richtig. Der andere molekulare Mechanismus betrifft direkt den Methylierungsgrad der DNA. Hier sind Bereiche, die eigentlich methyliert sein sollten, nicht mehr methyliert und umgekehrt. Es sind also die Lesezeichen selbst falsch gesetzt. Für beide Veränderungen gibt es krankheitsrelevante Beispiele.
Bereits seit den 1980er Jahren ist eine Korrelation zwischen einer DNA-Hypomethylierung und der Entstehung von Krebs bekannt. Diese Hypomethylierung wurde beispielsweise in repetitiven Sequenzen wie dem LINE-Element L1 gefunden, das mit der Entstehung etlicher Tumore, z. B. der Brust, Lunge, Leber oder Blase assoziiert sein kann. Durch Hypomethylierung wird dieses repetitive Element mobilisiert und führt so zu einer Instabilität des Genoms, was dann wiederum die Tumorentstehung fördert (Abb. 3 D).
Neben einem Verlust von 20 bis 60% der Methylierungen, die man in Tumorgewebe beobachten kann, kann allerdings auch Hypermethylierung zur Entartung von Zellen führen. Wie kann man sich das wiederum vorstellen? Wie bereits erwähnt, finden sich im Promotorbereich CpG-Inseln, die nicht oder wenig methyliert sind und darüber die Expression des nachgeschalteten Gens ermöglichen. Werden nun die CpG-Inseln in der Nähe eines Tumorsuppressorgens hypermethyliert, wird das Tumorsuppressorgen nicht mehr exprimiert, und es kommt zur Entartung der Zelle [5]. Eine Demethylierung oder Methylierung kann also sowohl einen guten als auch einen schlechten Effekt haben, und man muss schon ganz genau hinsehen, an welcher Stelle sich gegebenenfalls die Methylgruppe befindet. Inzwischen haben viele Wissenschaftler schon genau hingesehen und die Daten in einer allgemein zugänglichen, recht umfangreichen Datenbank gesammelt (http://methycancer.psych.ac.cn [6]).
Neben der Tumorentstehung ist die DNA-Methylierung auch mit anderen Krankheitsbildern assoziiert. Besonders das Zentrale Nervensystem zeigt hier eine Vielzahl an Beispielen. Als eines der komplexesten Systeme im Menschen unterscheiden sich einerseits die Expressionsmuster der verschiedenen Regionen, andererseits aber auch die eines bestimmten Zelltyps in Abhängigkeit davon, wo die spezielle Zelle im ZNS lokalisiert ist. An dieser Feinregulation der Transkription sind bevorzugt epigenetische Faktoren beteiligt.
Eine Erkrankung, die immer wieder mit der DNA-Methylierung in Verbindung gebracht wird, ist das Rett-Syndrom, eine neurologische Entwicklungsstörung, die durch eine Mutation im sogenannten MeCP2-Gen ausgelöst wird. Dieses Gen codiert für das Methyl-CpG-bindende Protein 2, also ein Protein, das DNA-Methylierung erkennt, daran bindet und über die Interaktion mit anderen Proteinen die Expression nachgeschalteter Gene reguliert [7].
Änderungen im Methylierungsmuster der Promotorregionen beeinflussen z. B. die Expression des neuronalen Faktors Reelin und auch des wichtigen brain-derived neurotrophic factor (BDNF) und sind dadurch an der möglichen Ausbildung von Epilepsien beteiligt [7]. Der Wachstumsfaktor BDNF ist allerdings in viele Prozesse im Gehirn involviert, so dass die deregulierte Expression eventuell Ursache etlicher neuronaler Erkrankungen ist. In klinischen Studien wird deshalb mittlerweile gezielt nach dem Zusammenhang zwischen der DNA-Methylierung des BDNF-Promotors und z. B. schweren Depressionen oder Schizophrenie gesucht (siehe www.clinicaltrials.gov). Andere Gene, deren Expression durch Hypermethylierung reprimiert (NEP) oder durch Hypomethylierung aktiviert (PADI2) wird, werden der Alzheimer-Erkrankung bzw. der multiplen Sklerose zugeordnet. Aber auch bei Autoimmunerkrankungen wie rheumatoider Arthritis oder systemischer Lupus erythematodes hat man bereits reprimierte (DR3) und aktivierte (PRF1, CD70, CD154, AIM2) Gene identifiziert [3]. Und es werden sicherlich noch viele weitere Gene die Liste zukünftig erweitern.
Epigenetische "Prophylaxe"
Für viele Krankheitsbilder kennt man sehr konkrete Risikofaktoren: Wer raucht, erhöht das Risiko, an Lungenkrebs oder einem Herzinfarkt zu sterben.
Was aber, wenn man kein so offensichtliches Korrelat findet? Wenn jemand Lungenkrebs bekommt, ohne jemals geraucht zu haben? Mittlerweile geht man dazu über, weit in der Vergangenheit des Betroffenen zu suchen und wird zum Teil auch fündig: und zwar ganz am Anfang [8]! Es ist ja auch irgendwie einleuchtend: In der Embryonalentwicklung wirken sich Schädigungen des Erbguts ganz extrem aus. Und so ist es ja auch ganz normal, dass eine Frau zu Beginn der Schwangerschaft tunlichst darauf achtet, sich keiner mutagen wirkenden Strahlenbelastung oder Toxinen auszusetzen. Allerdings beeinflusst eben nicht nur eine konkrete Mutation in der Basenfolge der DNA die Entwicklung des Embryos, sondern auch epigenetische Marker, allen voran die DNA-Methylierung, deren subtile Verteilung die zeitliche Abfolge exprimierter und reprimierter Gene reguliert. Da die hier sehr wichtigen DNA-Cytosin-5-Methyltransferasen auf eine ausreichende Versorgung mit S-Adenosylmethionin angewiesen sind, entscheidet letztlich auch die ausreichende Versorgung mit Methionin und Folat, ob das DNA-Methylierungsmuster korrekt aufgebaut und weiterentwickelt werden kann (Abb. 5).
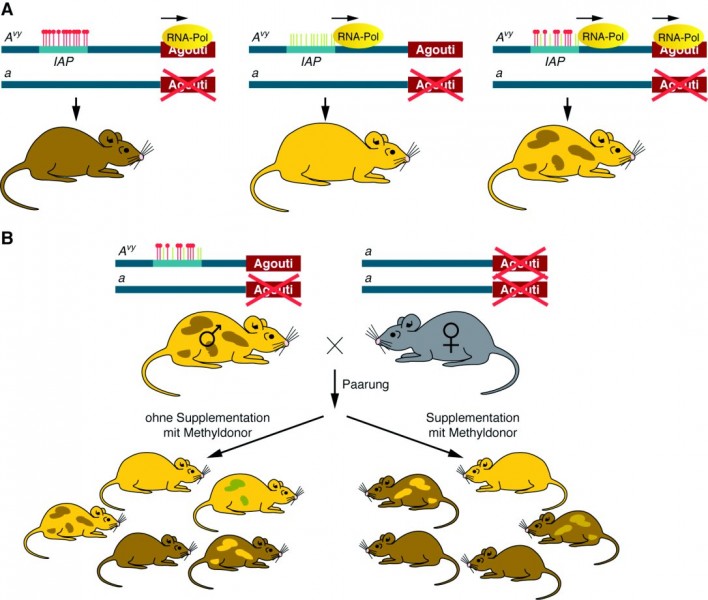
Dass die Ernährung bzw. auch die Supplementation mit Folsäure oder Vitamin B12 eine wichtige Rolle spielt, sieht man an den Honigbienen bzw. am sogenannten Agouti-Gen bei Mäusen. Die Larven der Honigbienen werden üblicherweise mit Pollen gefüttert und entwickeln sich ganz normal zu Arbeiterinnen. Nur wenn eine Larve Gelée royale erhält, schlüpft eine Königin. Der gleiche Effekt kann erzielt werden, wenn experimentell die Expression der DNA-Cytosin-5-Methyltransferase 3 inhibiert und damit das DNA-Methylierungsmuster beeinflusst wird [9].
Mäuse mit einer normalen Expression des Agouti-Gens sind braun und schlank. Bei einigen Mäusen findet sich jedoch das Agouti-Allel Avy (vy für viable yellow), wobei sich hier ein neues Steuerungselement in der Nähe des Gens befindet. Ist das Steuerungselement komplett methyliert, wird das Agouti-Gen normal exprimiert, und die Maus ist braun und schlank (Abb. 6 A). Bei einer kompletten Demethylierung ist die Agouti-Expression gestört und die Maus ist fettleibig und hat ein gelbes Fell. Ist das Steuerungselement teilweise methyliert, sind die Mäuse mehr oder weniger dick und haben ein braun geschecktes Fell. In einem Versuch wurden männliche Mäuse, deren zweites Agouti-Gen zerstört war und die deshalb nur das Avy -Gen exprimieren konnten, mit Weibchen gekreuzt, die überhaupt kein intaktes Agouti-Gen mehr hatten (Abb. 6 B). 50% der Nachkommen erben dabei rein statistisch den Avy /a-Genotyp und zeigen im Phänotyp den Methylierungsgrad des Steuerungselements IAP. Ein Teil der trächtigen Weibchen erhielt zusätzlich zum Futter noch einen Methylgruppendonor wie Folsäure oder Vitamin B12. Es zeigte sich, dass die Jungen der supplementierten Weibchen überwiegend braunes Fell hatten und schlank waren, während die Jungen der nicht-supplementierten Weibchen meist gelb und fettleibig waren. Allerdings gab es auch jeweils Junge mit einem gemischten bzw. dem jeweils entgegengesetzten Phänotyp. Dieses Experiment zeigt zweierlei: Zum einen, dass sich die Nährstoffversorgung während der Schwangerschaft direkt auf den Phänotyp der Nachkommen auswirken kann. Zum anderen zeigt es aber auch, dass genetisch an und für sich identische Nachkommen epigenetisch verschieden sein können. Das war auch bereits bei den Untersuchungen eineiiger Zwillinge beobachtet worden [10, 11].
Ein recht kompliziertes Bild, das sich da bezüglich des Methyloms der DNA auftut! Und es wird nicht einfacher, wenn man realisiert, dass Methylierungsmuster von den Eltern auch auf die Kinder und Enkelkinder weitervererbt werden können [10] und dass das Methylom tagtäglich z. B. von der Ernährung beeinflusst wird [11].
Kann/soll man also nun seine Ernährungsgewohnheiten und den Lebensstil ändern, um sein Methylom positiv zu beeinflussen? Wenn man sich nicht ausgewogen und in vernünftigem Maße ernährt und einen ungesunden Lebenswandel führt, dann ja, aber aus generellen gesundheitlichen Überlegungen. Aber gezielt im Übermaß Methylgruppendonoren zu supplementieren, um die DNA-Methylierung zu verstärken, könnte sich gerade z. B. bei Tumorsuppressorgenen, die ja nicht hypermethyliert werden dürfen, als Schuss nach hinten erweisen.
Therapie
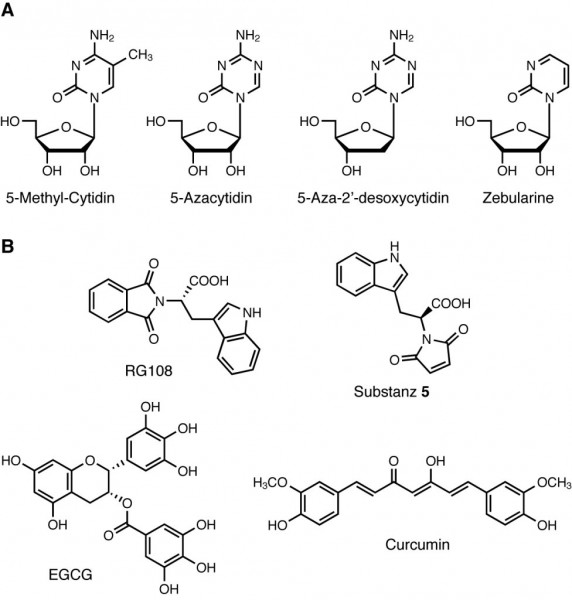
So wie kausale Zusammenhänge zwischen molekularen Mechanismen und einer Erkrankung – und hier vor allem bei einer Tumorerkrankung – gefunden werden, wird auch versucht, daraus Therapieansätze zu generieren. Und so ist es nicht verwunderlich, dass auch gezielt nach Inhibitoren der DNA-Cytosin-5-Methyltransferase DNMT1 gesucht wurde (Abb. 7). Seit 2008 ist in Europa als erster nukleosidischer Inhibitor Azacitidin (Vidaza®) zur Behandlung erwachsener Patienten zugelassen, die für eine Transplantation hämatopoetischer Stammzellen nicht geeignet sind und entweder an einem myelodysplastischen Syndrom mit intermediärem Risiko 2, an einer chronisch myelomonozytären Leukämie mit 10 bis 29% Knochenmarkblasten oder an einer akut-myeloischen Leukämie mit 20 bis 30% Blasten und Mehrlinien-Dysplasie leiden. Azacitidin wird in der Zelle über mehrere Schritte zu 5-Azacitidin-triphosphat (5-aza-CTP) phosphoryliert oder aber zu 5-Aza-desoxycytidin-triphosphat (5-aza-dCTP) metabolisiert. Man geht davon aus, dass ca. 80 bis 90% des verabreichten Azacitidins in Form von 5-aza-CTP in tRNAs und rRNAs der Zelle eingebaut wird. Nur 10 bis 20% gelangen in die DNA und können dort – falls sie in ein passendes CpG-Dinukleotid integriert werden – über eine irreversible Adduktbildung das DNMT1-Enzym inaktivieren [12]. Allerdings ist bei diesem Ansatz unklar, inwieweit die antineoplastische Wirkung von Azacitidin nicht auch auf andere zytotoxische Mechanismen wie z. B. der Hemmung der DNA-, RNA- und Proteinsynthese infolge des Einbaus des Nukleosid-Analogons in RNA und DNA zurückzuführen ist [13].
Neben Azacitidin ist von der FDA auch Decitabin (DacogenTM) zugelassen. Dieses Nukleosid-Analogon hat im Gegensatz zu Azacitidin bereits eine 2‘-Desoxyribose und wird nach der Phosphorylierung quantitativ in die DNA eingebaut [12]. Aber auch hier gilt, dass noch andere zytotoxische Effekte an der Wirksamkeit der Substanz beteiligt sein können. Klar ist, dass diese Nukleosid-Analoga, wie auch das ebenfalls schon lang beforschte Zebularine, starke Nebenwirkungen haben. Deshalb versucht man inzwischen, nicht-nukleosidische Inhibitoren zu entwickeln. Ausgehend von dem am Deutschen Krebsforschungszentrum (DKFZ) entwickelten Inhibitor RG108 wurden mittlerweile andere, zum Teil potentere nicht-nukleosidische Hemmstoffe der DNMT1 synthetisiert (Substanz 5, Abb. 7, [14]). Und es wurden weitere Kandidaten wie das Antihypertonikum Hydralazin oder das Antiarrhythmikum Procainamid sowie die Naturstoffe (–)-Epigallocatechin-3-gallat (EGCG) aus grünem Tee oder Curcumin aus der Gelbwurz als DNMT1-Hemmstoffe identifiziert [15]. Allerdings waren das alles Tests im Reagenzglas, und man darf gespannt sein, ob und wie sich die Substanzen in vivo bewähren.
Histon-Modifikationen …
Die Verpackung der DNA innerhalb des Zellkerns ist hoch komplex und doch auch ungleichmäßig. Nur das relativ locker gepackte Euchromatin steht für die Transkription zur Verfügung. Demgegenüber ist das stark kondensierte Heterochromatin wichtig für die Aufrechterhaltung der Chromosomenenden und für die korrekte Trennung der Chromatiden bei der Mitose.
Tab. 1: Histon-Modifikationen und assoziierte Funktionen [18] | ||
Modifikation |
modifizierte Aminosäure (Einbuchstaben-Code) |
Funktion |
Acetylierung |
K-ac |
Transkription, DNA-Reparatur, Replikation, Chromatin-Kondensation |
Methylierung (Lysin) |
K-me1, K-me2, K-me3 |
Transkription, DNA-Reparatur |
Methylierung (Arginin) |
R-me1, R-me2a, R-me2s |
Transkription |
Phosphorylierung |
S-ph, T-ph |
Transkription, DNA-Reparatur, Chromatin-Kondensation |
Ubiquitinylierung |
K-ub |
Transkription, DNA-Reparatur |
SUMOylierung |
K-su |
Transkription |
ADP-Ribosylierung |
E-ar |
Transkription |
Deiminierung |
R > Cit |
Transkription |
Prolin-Isomerisierung |
P-cis > P-trans |
Transkription |
Cit: Citrullin
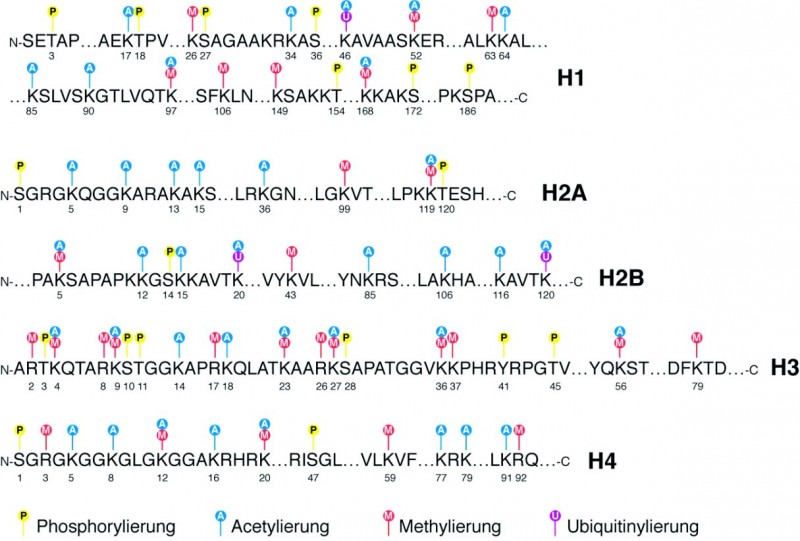
Ein wichtiger Mechanismus, der die Bildung von Heterochromatin bzw. Euchromatin beeinflusst, sind die Modifikationen der Histonproteine. Alle Histone des Nucleosoms können posttranslational modifiziert werden, wobei die wichtigsten Modifikationen Acetylierungen, Methylierungen, Phosphorylierungen, Ubiquitinylierungen, SUMOylierungen und ADP-Ribosylierungen sind (Abb. 8). Durch Acetylierung und Phosphorylierung werden die Ladungen der Nucleosomen und damit auch Abstoßungskräfte zwischen den Partikeln geändert. Das Anhängen des 76 Aminosäuren langen Proteins Ubiquitin oder des knapp 100 Aminosäuren langen Ubiquitin-ähnlichen Proteins SUMO (small ubiquitin-related modifier) an ein Histon ist eine sehr viel größere Modifikation. Dadurch können sterische Beeinträchtigungen der Nucleosomen-Verpackungseinheiten resultieren. Zusätzlich können aber auch spezielle Proteinkomplexe mit den kovalent verknüpften Gruppen bzw. Proteinen interagieren und für bestimmte Effekte, beispielsweise Transkription, Replikation oder auch DNA-Reparatur sorgen (Tab. 1). Insofern spricht man mittlerweile auch von einem "Histon-Code", der die Funktionen in einer Zelle beeinflusst. Inzwischen weiß man, dass beispielsweise Acetylierungen und Phosphorylierungen generell die Transkription fördern, während z. B. SUMOylierungen mit nicht-transkribierten Bereichen assoziiert sind.
Histon-Modifikationen – BegriffserklärungDie verschiedenen Histon-Modifikationen werden mit bestimmten Abkürzungen bezeichnet:
|
Demgegenüber findet man Methylierungen und Ubiquitinylierungen sowohl bei aktivierten als auch bei reprimierten Genen [16]. All diese Modifikationen sind enzymatisch katalysiert, und in den meisten Fällen gibt es auch Enzyme, die die Modifikationen wieder rückgängig machen. So sind z. B. etliche Histon-Acetylasen (HAT) und Histon-Desacetylasen (HDAC) für die Acetylierungen/Deacetylierung zuständig, Prolin-Isomerasen für die Isomerisierung von Prolin, Ubiquitilasen für die Ubiquitinylierung und Methyltransferasen sowie Demethylasen für die Methylierung/Demethylierung. Erstaunlicherweise sind gerade die Methyltransferasen und Demethylasen extrem spezifisch, indem sie ganz bestimmte Aminosäuren methylieren.
Genomic Imprinting
oder der Unterschied zwischen Maultier und Maulesel
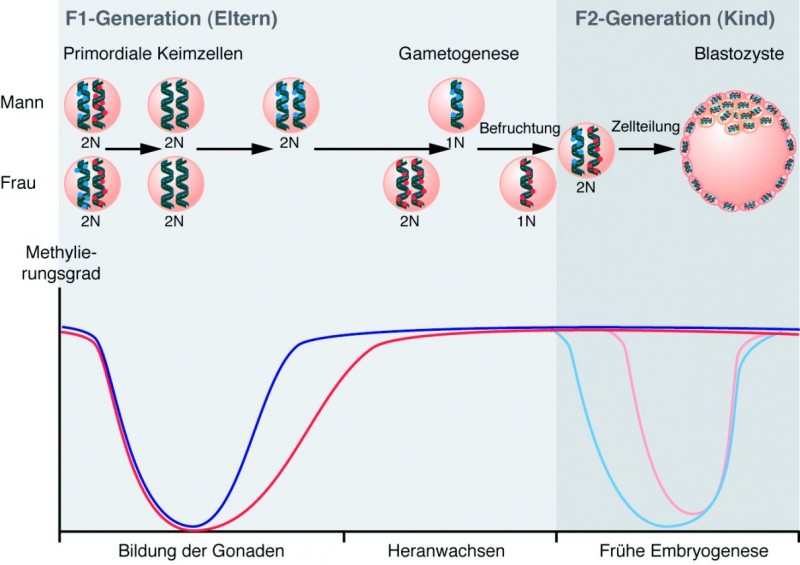
Maultier und Maulesel gelten als DIE Beispiele schlechthin, dass ein Organismus eben nicht aus der Summe seiner Gene resultiert: In beiden Fällen kommt das Erbmaterial von Esel und Pferd zusammen und doch entstehen recht unterschiedliche Tiere! Maulesel haben eine Esel-Mutter und einen Pferd-Vater, während Maultiere von einer Pferd-Mutter und einem Esel-Vater abstammen; dabei ähneln die resultierenden "Kinder" jeweils sehr viel stärker der Mutter als dem Vater. Eine Erklärung dafür ist, dass die Eizelle der Mutter den Großteil des Zytoplasmas und der Zellorganellen der späteren Zygote bereitstellt. Die andere Erklärung liegt in einer aufwendigen genetischen Reprogrammierung der Keimzellen bezüglich ihrer DNA-Methylierung: Zunächst werden die Genome in den primordialen Keimzellen komplett demethyliert. Mit der Reifung der Gameten werden wieder Methylgruppen platziert, die dann allerdings nach der Befruchtung erneut entfernt werden. Dabei wird das väterliche Genom aktiv und komplett demethyliert, während die bestehende Methylierung des mütterlichen Genoms dadurch "ausgedünnt" wird, dass der neu synthetisierte DNA-Strang nach der Replikation nicht wieder methyliert wird. Bestimmte Methylierungsmarken in sogenannten geprägten (imprinted) Genen bleiben jedoch während dieses Vorgangs erhalten. Dadurch ist die spätere Expression derartiger Gene im Organismus unterschiedlich, je nachdem ob das Gen ursprünglich vom Vater oder von der Mutter stammte. Mit der Einnistung des Embryos in die Gebärmutter wird die DNA erneut umfassend de novo methyliert, um dann die Genexpression der unterschiedlichen Körperzellen gezielt zu steuern (siehe Abb. "Genomic Imprinting").
Man geht inzwischen davon aus, dass ca. 1% der Gene in Säugetieren während der Embryogenese derartig geprägt ist. Im menschlichen Genom hat man über 150 mögliche, imprinted Gene identifiziert, die also das ursprüngliche Keimbahnmuster und die von den Eltern geerbte Aktivität während der ganzen Entwicklung beibehalten. Fehler im "genomic imprinting" können zu schweren Krankheitsbildern führen, wie zu dem Prader-Willi-Syndrom, dem Angelman-Syndrom, dem Silver-Russell-Syndrom oder aber dem Beckwith-Wiedemann-Syndrom.
Bei assistierten Reproduktionstechniken, also beispielsweise der In-vitro-Fertilisation, kann es durch den Eingriff in die normale Gameten- und Embryonalentwicklung ebenfalls zu Störungen des "genomic imprinting" und damit zu einem leicht gehäuften Auftreten der erwähnten Krankheiten kommen.
Und nicht zuletzt sind etliche Schwierigkeiten beim Transfer des Zellkerns einer somatischen Zelle in eine entkernte Eizelle mit dem Ziel, einen Organismus zu klonen, auf die fehlende Reprogrammierung der DNA-Methylierung zurückzuführen.
Quelle
Butler, M.G.: Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet 26:477 – 486 (2009) Jirtle, R.L., Skinner, M.K.: Environmental epigenomics and disease susceptibility. Nat Rev Genet. 8: 253 – 62 (2007).
… und ihre Bedeutung im gesunden …
Euchromatin ist üblicherweise dadurch charakterisiert, dass die Histone insgesamt stark acetyliert sind (Abb. 9) und das Histon H3 an den Lysinen (K) an den Aminosäure-Positionen 4, 36 und 79 trimethyliert ist. Demgegenüber zeichnet sich Heterochromatin durch eine geringe Acetylierung und eine Methylierung an H3K9, H3K27 und H4K20 aus [3].
Das sind also bekannte Modifikationen, die die Chromatinstruktur beeinflussen. Auf Gen-Ebene kennt man ebenfalls spezifische Veränderungen an den Histonen: Für die Transkriptionsinitiation scheinen H3K27ac, H2BK5ac, H3K4me3 und H4K20me1 wichtig zu sein, während H3K79me1 und H4K20me1 für die Weiterführung der Transkription über das ganze Gen sorgen.
Reichlich kompliziert dieser Histon-Code! Und es wird noch komplizierter, weil offensichtlich auch eine Präferenz zwischen bestimmten Histon-Modifikationen und Promotoren mit hohem CpG-Anteil und solchen mit geringerem CpG-Anteil vorhanden ist [16]. Man kann also keineswegs DNA-Methylierung und Histon-Modifikation getrennt voneinander betrachten. Vielmehr beeinflussen sie sich gegenseitig, so dass beispielsweise eine DNA-Methylierung auch eine Methylierung an H3K9 bedingt. Und dann gibt es noch die Histon-Modifikationen, die nur dann angehängt werden, wenn das Histon bereits an anderer Stelle mit einer bestimmten Modifikation markiert ist [16].
Im Unterschied zur DNA-Methylierung scheint es keine Vererbung des Histon-Codes zu geben, so dass die Histon-Modifikationen strenggenommen nicht der eigentlichen Begriffserklärung der "Epigenetik" entsprechen. Dennoch sind sie ganz klar mit einer Reihe epigenetischer Phänomene assoziiert [17].
Wie kompliziert das Zusammenspiel der epigenetischen Modifikationen ist, sieht man daran, dass in einer Modellrechnung unter Berücksichtigung der verschiedenen funktionellen Elemente allein 51 unterschiedliche Chromatin-Formen in T-Zellen identifiziert wurden [18]. Anfang des Jahres hat sich ein internationales Konsortium zusammengefunden, das sich zur Aufgabe gemacht hat, innerhalb der nächsten 10 Jahre 1000 epigenetische "Fingerabdrücke" von jedem Zelltyp des Menschen anzufertigen. Eine Fülle von Daten hat sich bereits beispielsweise beim Epigenome Roadmap Project (http://www.roadmapepigenomics.org) angesammelt.
… und im kranken Organismus
Die verschiedenen Modifikationen, und hier vor allem Acetylierungen und Methylierungen der Histone, beeinflussen die Chromatinstruktur und die Transkription von Genen. Kommt es zu unkontrollierten Modifikationen oder zu irregulären Desacetylierungen und Demethylierungen, können daraus Krankheiten entstehen. Die erste Korrelation konnte man zwischen einer allgemeinen Desacetylierung, v. a. an Position H4K16, mit Tumoren herstellen. Derartige Änderungen können unterschiedliche Ursachen haben: Zum einen können die modifizierenden Enzyme mutiert sein, zum anderen können die de-modifizierenden Enzyme mutiert sein, und zum dritten können auch wieder Proteine mutiert sein, die normalerweise die Modifikation "lesen" und die Gen-Transkription steuern. In etlichen Tumoren finden sich überexprimierte oder mutierte Histon-Desacetylasen, was dann zu einer verstärkten Desacetylierung oder aber zu einem geänderten Acetylierungsmuster führt. Und wenn Tumorsuppressorgene betroffen sind, werden die nicht mehr transkribiert, und es kann zu einer Zellentartung kommen.
Von den bekannten Lysin-Methylierungen ist ein Verlust der aktiven Markierung H3K4me3 oder auch der repressiven Markierung H4K20me3 mit Krebszellen assoziiert. Daneben können auch die repressiven Modifikationen H3K9me und H3K27me3 zusätzlich auftreten und ebenfalls zu einer Tumorentstehung führen. Und auch hier sind zum Teil die Methylasen oder auch Demethylasen mutiert oder überexprimiert.
Neben Tumorerkrankungen kennt man auch weitere Krankheitsbilder, die mit veränderten Histon-Modifikationen einhergehen. So ist z. B. das Rubinstein-Taybi-Syndrom eine mit einer Häufigkeit von 1:120.000 autosomal dominant auftretende Erkrankung, die durch eine Dysfunktion einer Histon-Acetyltransferase hervorgerufen wird. Dadurch kommt es zu Kleinwuchs und einer unterschiedlich stark ausgeprägten geistigen Behinderung. Hypoacetylierungen der Histone sieht man auch bei der amyotrophen Lateralsklerose, bei Morbus Parkinson, Chorea Huntington oder der Friedreich-Ataxie. Bei den letztgenannten Krankheitsbildern tritt zusätzlich auch noch eine Hypertrimethylierung an H3K9 auf. Hyperacetylierungen werden allerdings ebenfalls mit Erkrankungen in Verbindung gebracht. Entsprechende Aktivierungen hat man z. B. im Promotorbereich von Entzündungsgenen gefunden, die dann z. B. zu Diabetes oder Asthma führen können.
Therapie
Nachdem Hypoacetylierungen sehr häufig mit Tumorerkrankungen assoziiert vorliegen, war es naheliegend, Inhibitoren der Histon-Desacetylasen als Therapeutika zu entwickeln. Die ersten Wirkstoffe, Vorinostat/SAHA (Zolinza®) und Romidepsin (ISTODAX®) sind bereits von der FDA für die Behandlung kutaner T-Zell-Lymphome (CTCL) zugelassen. Weitere Substanzen sind in fortgeschrittener klinischer Entwicklung (Abb. 10).
Histon-Desacetylasen lassen sich in vier Klassen einteilen. Die Enzyme der Klassen I, II und IV katalysieren Zink-abhängig die Amidspaltung, und man hat beobachtet, dass Butyrat, Diallylsulfid und Sulforaphan aus der Nahrung die HDACs der Klasse I und II inhibieren können. HDACs der Klasse III werden auch als Sirtuine bezeichnet und benötigen NAD+ zur Amidspaltung. Hier zeigte es sich, dass sich diese Enzyme z. B. durch Resveratrol aktivieren lassen [19]. Über den einzigen Vertreter der Klasse IV, die HDAC 11, ist bisher noch wenig bekannt. Die in Abbildung 10 gezeigten Inhibitoren richten sich gegen die Zink-abhängigen HDACs der Klassen I, II und IV. In verschiedenen Untersuchungen hat sich gezeigt, dass die Hauptwirkungen der HDAC-Inhibitoren darin liegen, den Zellzyklus anzuhalten, Zelldifferenzierung zu induzieren und Apoptose weiterzuführen. Darüber hinaus können die Substanzen die Sensitivität der Tumorzellen gegen eine Chemotherapie erhöhen und die Angiogenese inhibieren [20].
Sirtuine haben eine wichtige Rolle in vielen zellulären Prozessen wie z. B. das Abschalten von Genen, die Regulation der Transkriptionsfaktoren, die Regulation des Zellzyklus, den Fettsäuremetabolismus sowie die Lebenszeitverlängerung. Deshalb unterscheiden sich die verschiedenen Sirtuine hinsichtlich der zellulären Lokalisation, der Substratspezifität und ihrer biologischen Funktionen. Der am besten charakterisierte Vertreter ist SIRT1. SIRT1 ist im Zellkern lokalisiert und ist beispielsweise in Tumorzellen von Lungenkrebs, Prostatakrebs und Leukämien verstärkt exprimiert. Als physiologischer Inhibitor der Sirtuine gilt Nicotinamid; die ersten synthetischen Inhibitoren sind Sirtinol und Splitomicin (Abb. 11). Ob diese Moleküle in der oder ähnlicher Form jedoch irgendwann als Tumortherapeutika zur Verfügung stehen werden, muss die Zukunft zeigen.
Genom-Organisation
Alle Mechanismen, die die Ausbildung von Heterochromatin verstärken, beeinflussen also auch die Expression von Genen. Neben den bereits diskutierten epigenetischen Modifikationen lernt man immer wieder neue Interaktionen kennen, die die Genom-Organisation beeinflussen [21]. Relativ neu sind die Erkenntnisse, dass RNAs hier ebenfalls mitmischen. Zum einen handelt es sich um die bereits bekannten Antisense-RNAs, also RNAs mit einer komplementären Sequenz zu mRNAs der Zelle. Bislang war man davon ausgegangen, dass eine Antisense-RNA im Zytoplasma der Zelle mit ihrer mRNA einen Doppelstrang ausbildet und darüber die Bildung des Proteins steuert. Nun hat man gefunden, dass sich die Antisense-RNA im Zellkern an der Bildung des Heterochromatins beteiligt und so eventuell sogar über weite Genombereiche die Expression mehrerer Gene steuert. Eine andere Gruppe von RNAs sind die lincRNAs, die als "large intergenic non-coding" RNAs bezeichnet werden, also RNAs, die von DNA-Bereichen transkribiert werden, die zwischen den eigentlichen Genen liegen und eben keine Information für ein Protein enthalten. Man geht inzwischen davon aus, dass aus dem menschlichen Genom ca. 4500 solcher lincRNAs generiert werden [22]. Die Funktion all dieser Moleküle ist noch nicht komplett aufgeklärt. Sicher ist jedoch, dass sie mindestens zum Teil an der Bildung des Heterochromatins beteiligt sind und über die Interaktion mit entsprechenden Proteinkomplexen die Methylierung der Histone steuern.
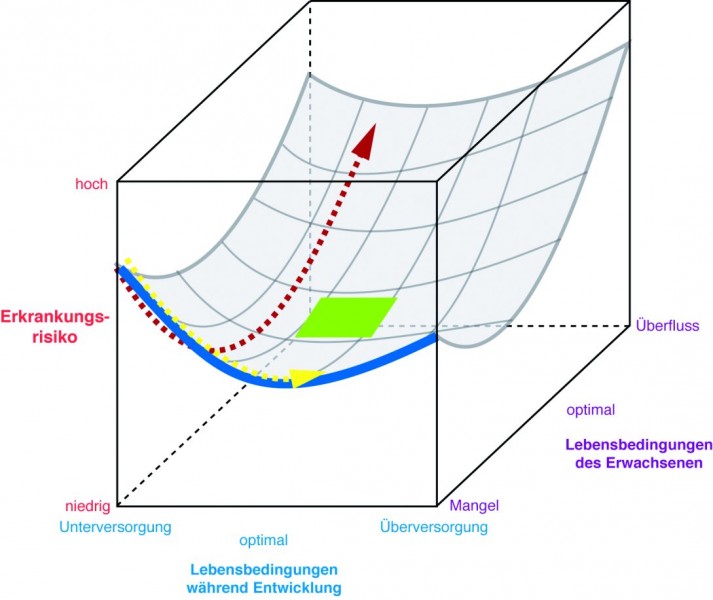
Man darf gespannt sein, wie sich das weite Feld der Epigenetik weiter entwickeln wird. Das Epigenom scheint auf jeden Fall sehr viel komplexer zu sein, als die simple Abfolge von Basenpaaren, die die Basis der genomischen Codierung bildet. Interessant sind die Untersuchungen, die sich mit der "Developmental Origin of Health and Disease" -Hypothese befassen. Nach dieser Hypothese wird unser Epigenom während der Entwicklung in der Gebärmutter und auch während der Stillzeit von Stress, Nahrungsangebot und eventuellen teratogenen Substanzen entscheidend geprägt und auf die momentanen Lebensbedingungen der Mutter eingestellt (Abb. 12). Je stärker sich diese Lebensumstände dann allerdings von den Lebensumständen des heranwachsenden Kindes unterscheiden, desto größer ist das Erkrankungsrisiko für das Kind. Konkret: Ist der Fetus unterernährt, weil die Mutter nicht genügend Essen zur Verfügung hatte, stellt sich das Epigenom so ein, dass Gene exprimiert werden, die das vorhandene Nahrungsangebot optimal ausschöpfen. Schöpft das Kind dann später aber aus einem reichlichen Nahrungsangebot, ist die Gefahr sehr groß, an Typ-2-Diabetes und Herz-Kreislaufkomplikationen zu erkranken [23].
Epigenetisch bedingte Frust-Schokolade
Wer sich zum Jahresbeginn wieder vorgenommen hat, den angefutterten Weihnachtsspeck durch eine Diät loszuwerden, kann sich das eventuell sparen – die Epigenetik könnte dagegen sprechen.
Hinweise darauf kamen von Studien an männlichen Mäusen [1]. Diese Mäuse wurden einer dreiwöchigen Kalorienreduktion auf 75% unterzogen, was zu einem Gewichtsverlust von 10 bis 15% führte. Dieser Wert wird auch beim Menschen bei einer erfolgreichen Diät erreicht. Im Stresstest zeigte sich, dass die abgemagerten Mäuse wesentlich stärker reagierten als ihre normal gefütterten Artgenossen, dass sie deutlich erhöhte Corticosteroid-Konzentrationen aufwiesen und länger brauchten, um sich von der Stresssituation zu erholen. Und wenn man den Mäusen nach einer Stresssituation zusätzlich zur normalen Versorgung fettreiches Futter anbot, neigten die abgemagerten Mäuse eher dazu, die tägliche Kalorienzufuhr über fettreiche Nahrung zu bestreiten, als die Kontrollmäuse.
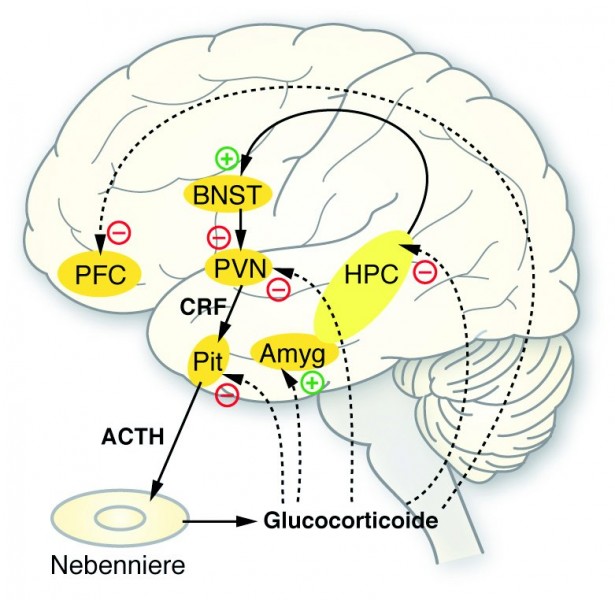
Molekulare Untersuchungen brachten dann den zugrundeliegenden Mechanismus ans Licht: Durch die Diät wurde der Grad der DNA-Methylierung in den CpG-Inseln im Bereich des Promotors für den corticotropin releasing factor (CRF) verringert, was in diesem Fall zu einer verminderten Expression von CRF führte. Allerdings nur im sogenannten Bed nucleus der Stria terminalis (BNST). Demgegenüber blieb die DNA-Methylierung und auch die CRF-Expression im zentralen Nucleus der Amygdala gleich. Was bedeutet das?
GABAerge Neurone, die CRF enthalten, inhibieren den paraventrikulären Nucleus im Hypothalamus (PVN). PVN wiederum regt mit CRF die Hypophyse (Pit) zur Produktion von ACTH (adrenocorticotropes Hormon) an, was dann die Freisetzung von Glucocorticoiden aus den Nebennieren induziert [2]. Verringert sich also die CRF-Bildung im BNST, wird letztlich die Glucocorticoid-Bildung gesteigert (Abb. "Zentrale Rolle des CRF".). Durch diese Steigerung des Stresssignalwegs wird auch das Belohnungssystem des Gehirns stimuliert, weshalb die gestressten Mäuse bevorzugt die fettreiche Nahrung zu sich nahmen – quasi als "Droge", um mit der Stresssituation besser fertig werden zu können. Molekular konnte das Frustessen über die Expression des Melanin-concentrating-Hormons (MCH) bzw. des Orexins nachgewiesen werden. Nur bei den Mäusen, die vor der fettreichen Nahrung kalorienreduziert gehalten wurden, war die Expression der beiden Hormone erhöht, nicht jedoch bei denjenigen, die die ganze Zeit normal fressen konnten. Beide Hormone greifen in das mesolimbische dopaminerge System ein und steigern die Motivation, hochkalorische Nahrung aufzunehmen.
Die epigenetische Modifikation des CRF-Promotors war wahrscheinlich evolutionär wichtig, als Nahrung noch knapp und die Nahrungssuche relativ stressig war. Mit der heutigen hochkalorischen Ernährungsweise führt diese Modifikation dann aber eher zu krank machendem Übergewicht. Und wenn die Kalorienreduktion nachhaltig zur DNA-Demethylierung des CRF-Promotors führt, kann man sich die Diät eigentlich schenken, schließlich würde man dadurch die Stresskaskade verstärken. Das ist sicherlich eine gute Ausrede für all diejenigen, die eigentlich gar nicht abnehmen wollen. Alle anderen seien daran erinnert, dass wir uns im Unterschied zu den Mäusen mit unserer Willenskraft auch aktiv gegen die Frust-Schokolade entscheiden und auf diese Art abnehmen können.
Vielleicht bieten sich aber mit diesen neuen Erkenntnissen ganz interessante Ansatzpunkte, um mit geeigneten Inhibitoren eine langfristig erfolgreiche Diät zu unterstützen.
[1] Pankevich, D.E., Teegarden, S.L., Hedin, A.D., et al.: Caloric Restriction Experience Reprograms Stress and Orexigenic Pathways and Promotes Binge Eating. J Neurosc. 30 (2010) 16399 – 16407.
[2] Koob, G.F., Le Moal, M.: Drug Addiction, Dysregulation of Reward, and Allostasis. Neuropsychopharmacol. 24 (2001) 97 – 128.
Ist jetzt also nach den Genen auch noch das Epigenom daran Schuld, wenn die Bevölkerung zu dick ist? Sicherlich entscheidet eine epigenetische Prägung über die Entwicklung des Fetus und letztlich auch über die Anfälligkeit gegenüber Krankheiten. Ob jedoch wirklich derzeit ein kausaler Zusammenhang zwischen einer bestimmten DNA-Methylierung oder einer bestimmten Histon-Modifikation und beispielsweise einem späteren Bluthochdruck hergestellt werden kann, ist sehr fraglich: Zu komplex und noch zu wenig geklärt ist das Zusammenspiel zwischen den verschiedenen epigenetischen Markern zu einem bestimmten Zeitpunkt in einer bestimmten Zelle [24]. Und ob sich dann irgendwann irgendwelche Empfehlungen ableiten lassen, wie man sich als Schwangere zu ernähren und zu verhalten hat, damit das Kind später einmal gesund durchs Leben gehen kann, bleibt abzuwarten. Zunächst einmal wird sich zeigen, inwieweit sich therapeutische Eingriffe in das Epigenom bei Tumorerkrankungen bewähren, oder aber ob dabei doch zu viele Lesezeichen verändert werden.
Literatur
[1] Van Speybroeck, L.: From Epigenesis to Epigenetics – The Case of C. H. Waddington. Ann. N.Y. Acad. Sci. 981: 61– 81 (2002).
[2] Suzuki, M.M., Bird, A.: DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Gen. 9: 465 – 467 (2008)
[3] Portela, A., Esteller, M.: Epigenetic modifications and human disease. Nat. Biotechn. 28: 1057– 1068 (2010)
[4] Fraga, M.F., Ballestar, E., Paz, M.F.: Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 102:10604 – 10609 (2005).
[5] Robertson, K.D.: DNA methylation and human disease. Nat. Rev. Gen. 6: 597 – 610 (2005)
[6] He, X., Chang, S., Zhang, J.: MethyCancer: the database of human DNA methylation and cancer. Nucleic Acids Res. 36: D836 – D841 (2008)
[7] Qureshi, I.A., Mehler, M.F.: Epigenetic mechanisms underlying human epileptic disorders and the process of epileptogenesis. Neurobiol Dis. 39:53 – 60 (2010)
[8] Gheorghe, C.P., Goyal, R., Mittal, A., Longo, L.D.: Gene expression in the placenta: maternal stress and epigenetic responses. Int J Dev Biol. 54:507 – 23 (2010)
[9] Kucharski, R., Maleszka, J., Foret, S., Maleszka, R.: Nutritional Control of Reproductive Status in Honeybees via DNA Methylation. Science 28: 1827 – 1830 (2008)
[10] Jirtle, R.L., Skinner, M.K.: Environmental epigenomics and disease susceptibility. Nat Rev Genet. 8: 253 – 62 (2007).
[11] Gallou-Kabani, C., Vigé, A., Gross, M.S., Junien, C.: Nutri-epigenomics: lifelong remodelling of our epigenomes by nutritional and metabolic factors and beyond. Clin Chem Lab Med. 45: 321 – 327 (2007).
[12] Stresemann, C., Lyko, F.: Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 123: 8 – 13 (2008).
[13] EPAR-Produktinformation Vidaza® , http://www.ema.europa.eu
[14] Suzuki, T., Tanaka, R., Hamada, S., Nakagawa, H., Miyata, N.: Design, synthesis, inhibitory activity, and binding mode study of novel DNA methyltransferase 1 inhibitors. Bioorg Med Chem Lett. 20: 1124 – 1127 (2010).
[15] Kuck, D., Singh, N., Lyko, F., Medina-Franco, J.L.: Novel and selective DNA methyltransferase inhibitors: Docking-based virtual screening and experimental evaluation. Bioorg Med Chem 18: 822 – 829 (2010).
[16] Karlic´, R., Chung, H.R., Lasserre, J., Vlahovicek, K., Vingron, M.: Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 107: 2926 – 2931 (2010).
[17] Kouzarides, T.: Chromatin Modifications and Their Function. Cell 128: 693 – 705 (2007)
[18] Ernst, J., Kellis, M.: Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechn. 28: 817 – 825 (2010)
[19] Davis, C.D., Ross, S.A.: Dietary Components Impact Histone Modifications and Cancer Risk. Nutr. Rev. 65: 88 – 94 (2007).
[20] Mai, A., Altucci, L.: Epi-drugs to fight cancer: From chemistry to cancer treatment, the road ahead. Int J Biochem Cell Biol. 41: 199 – 213 (2009)
[21] Feinberg, A.P.: Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat. Biotechn. 28: 1049 – 1052 (2010)
[22] Khalil, A.M., Guttman, M., Huarte, M., et al.: Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 106: 11667 – 11672 (2009)
[23] Godfrey, K.M., Gluckman, P.D., Hanson, M.A.: Developmental origins of metabolic disease: life course and intergenerational perspectives. Trends Endocrinol Metab. 21: 199 – 205 (2010)
[24] Attig, L., Gabory, A., Junien, C.: Early nutrition and epigenetic programming: chasing shadows. Curr Opin Clin Nutr Metab Care. 13: 284 – 293 (2010).
Autoren
Dr. Ilse Zündorf, Prof. Dr. Theodor Dingermann, Institut für Pharmazeutische Biologie, Biozentrum, Max-von-Laue-Str. 9, 60438 Frankfurt/Main
Zum Weiterlesen
Inhibitoren von Histon-Desacetylasen und DNA-Methyltransferasen
Pharmazie in unserer Zeit Ausgabe 3/10
Gastherausgeber: Prof. Dr. Theo Dingermann, Prof. Dr. Manfred Jung, Prof. Dr. Wolfgang Sippl
Die Zeitschrift "Pharmazie in unserer Zeit" wird von der Deutschen Pharmazeutischen Gesellschaft e.V. (DPhG) herausgegeben. Mitglieder der DPhG erhalten die Zeitschrift kostenlos. Weitere Informationen unter www.dphg.de.
Zellbiologie: Lesezeichen im Buch des Lebens – Epigenetik (Julia Mareike Wagner, Prof. Dr. Manfred Jung)
Medizinische Chemie: Entwicklung von Histon-Desacetylaseinhibitoren – Potente Wirkstoffe vor allem bei Krebserkrankungen (Ralf Heinke, Prof. Dr. Wolfgang Sippl)
Klinik: Vorinostat in der Behandlung kutaner T-Zell-Lymphome – Therapie mit Histon-Desacetylase-Inhibitoren (Dr. Katja Zirlik, Prof. Dr. Dorothée Nashan, Prof. Dr. Hendrik Veelken)
Klinik: Valproinsäure als Histon-Desacetylase-Hemmstoff – Neues Einsatzgebiet für einen altbekannten Arzneistoff (Julia Mareike Wagner, PD Dr. Gesine Bug, Prof. Dr. Manfred Jung)
Klinik: HDAC-Inhibitoren als Therapie für neuronale Erkrankungen – Entdeckung neuer Anwendungsgebiete (Dr. Andre Fischer)
Zellbiologie: Die Entzifferung der fünften DNA-Base – Das Methylom des Menschen (Prof. Dr. Thomas Winckler, Dr. Ilse Zündorf, Prof. Dr. Theo Dingermann)
Klinik: Epigenetische Therapie bei Myelodysplastischen Syndromen (MDS) – Therapie mit DNA-Methyltransferase-Inhibitoren (Dr. Michael Daskalakis, Dr. Nadja Blagitko-Dorfs, Dr. Björn Hackanson, Prof. Dr. Michael Lübbert)
Klinik: Supportive Therapie bei Myelodysplastischem Syndrom (MDS) – Unverzichtbare Säule im multimodalen Behandlungskonzept (Dr. Alexandra Göbel, Dr. Beate Lubrich)



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.