
















- DAZ.online
- DAZ / AZ
- DAZ 38/2008
- Wozu brauchen wir eine ...
Arzneimittelsicherheit
Wozu brauchen wir eine Arzneimittelkommission?
Deutsches Apothekerhaus in Eschborn
In diesem Gebäudekomplex ist auch die Arzneimittelkommission
der Deutschen Apotheker untergebracht.
Foto: ABDA
Im Jahr 1929 schrieb der damals bereits berühmte Pharmakologe Paul Trendelenburg in seinen "Grundlagen der allgemeinen und speziellen Arzneiverordnung", er wolle "durch Auswahl der wichtigen Mittel und Zurücktretenlassen des Unwichtigen oder noch nicht genügend Erprobten dazu beitragen, dass der werdende Arzt wieder in den Stand versetzt wird, besser zu beurteilen, wann er mit seinem therapeutischen Handeln auf festem Boden steht. Früher war der Arzneischatz etwas relativ Stabiles, und die Stimmen, die seinen therapeutischen Wert beurteilten, bemühten sich im Allgemeinen der Objektivität. Seit die Arzneimitteldarstellung fast ganz dem Kapitalismus unterworfen ist, erschwert die Unsumme immer neu auftauchender Spezialitäten und die oft recht subjektiv gehaltene Form ihrer Empfehlung die Bildung eines sicheren Urteils" [1].
Maßgeblich auf Betreiben des Pharmakologen Wolfgang Heubner in seiner Göttinger Zeit von 1908 bis 1928 und des Internisten Adolf Schmidt, ehemals Direktor der medizinischen Klinik Halle/Saale, wurde schon 1911 auf dem Wiesbadener Internistenkongress eine Kommission eingesetzt, die fortan "Arzneimittelkommission" hieß und eine Fachkommission der späteren Deutschen Gesellschaft für Innere Medizin war [2]. Erst 1951 ging aus der Arzneimittelkommission der Deutschen Gesellschaft für Innere Medizin dann die Arzneimittelkommission der deutschen Ärzteschaft hervor.
Heubner, der auch ein guter, durchsetzungsstarker Redner war, scheute im Interesse einer rationalen Therapie auch die direkte Auseinandersetzung nicht und führte schon in seinen jüngeren Jahren einen kompromisslosen Kampf mit Vertretern paramedizinischer, dogmatischer Therapierichtungen [3]. Auch sah er die Pharmakologie stets in der Pflicht zur ständigen Verbesserung der Arzneitherapiesicherheit und zu einer unabhängigen, von wirtschaftlichen Interessen freien Arzneimittelprüfung.
Die Anfänge
Fast 25 Jahre später, im Jahre 1975, wurde durch Beschluss des damaligen ABDA-Präsidiums unter Richard Fellmann, Walter Riemerschmid und Götz Alberti die Arzneimittelkommission der Deutschen Apotheker (AMK) gegründet und nahm unter ihrem ersten Vorsitzenden Heinz Glück und dem langjährigen unmittelbaren Nachfolger Volker Dinnendahl (seit 1978) die Arbeit auf. Auslöser der AMK-Gründung war die bevorstehende Verabschiedung des neuen Arzneimittelgesetzes (AMG) von 1976, dessen Konzeption als Richtschnur die Kriterien der Qualität, Wirksamkeit und Unbedenklichkeit von Arzneimitteln enthielt und maßgeblich durch die Contergan-Katastrophe von 1961 beeinflusst worden war. Heute ist schon fast vergessen, dass das alte Arzneimittelgesetz von 1961 gar kein Zulassungsverfahren für Arzneimittel definierte, sondern nur eine behördliche Herstellungserlaubnis und eine Registrierungspflicht beinhaltete.
Erst 1978 trat dann das neue Arzneimittelgesetz in Kraft, dessen Kern in der Einführung der behördlichen Zulassung der Arzneimittel bestand und das durch die Regelung und Vorgabe von verbindlichen klinischen Studien die dringend notwendige Antwort auf die Contergankatastrophe von 1961 war.
Erfassung und Auswertung von Arzneimittelrisiken gemäß AMG
Die Rechtsgrundlage für die Tätigkeit der AMK findet sich in § 62 AMG, der den Titel trägt "Beobachtung, Sammlung und Auswertung von Arzneimittelrisiken". Der Gesetzgeber versteht unter diesen Risiken insbesondere
- Nebenwirkungen,
- Wechselwirkungen mit anderen Mitteln,
- Verfälschungen sowie
- potenzielle Umweltrisiken aufgrund der Anwendung eines Tierarzneimittels.
Die zentrale Erfassung und Auswertung von Arzneimittelrisiken, die nicht aufgrund eines Mangels der pharmazeutischen Qualität entstehen, sowie die Koordination der zu ergreifenden Abwehrmaßnahmen obliegen den Zulassungsbehörden. Dies sind
- für zentral zugelassene Arzneimittel die Europäische Arzneimittelagentur EMEA (European Medicines Agency),
- das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) für den Zuständigkeitsbereich aller übrigen Arzneimittel und Medizinprodukte,
- das Paul-Ehrlich-Institut (PEI) für den Zuständigkeitsbereich biomedizinischer Arzneistoffe (z. B. Sera und Impfstoffe),
- das Bundesamt für Verbraucherschutz und Lebensmittelsicherheit (BVL) für den Zuständigkeitsbereich Tierarzneimittel.
Die Bundesoberbehörden arbeiten in der Verfolgung der Risikoabwehr nicht nur mit den für die Durchführung des Arzneimittelgesetzes zuständigen Behörden der Bundesländer zusammen, sondern auch mit internationalen Stellen wie der WHO und der EMEA sowie mit anderen nationalen Arzneimittelbehörden. Im Inland ist u. a. auch das Zusammenwirken mit den Arzneimittelkommissionen der Kammern der Heilberufe im Gesetz festgeschrieben.
Stufenplan bei Arzneimittelrisiken
Wie dieses Zusammenwirken der beteiligten Institutionen im Einzelnen abläuft und in welcher Weise die Öffentlichkeit durch die Bundesoberbehörde informiert wird, übrigens durchaus auch vor Abschluss des betreffenden Verfahrens, dann aber "ergebnisoffen", dieses Zusammenwirken ist in einer "Allgemeinen Verwaltungsvorschrift zur Beobachtung, Sammlung und Auswertung von Arzneimittelrisiken (Stufenplan) nach § 63 des Arzneimittelgesetzes" festgelegt. Dort sind u. a. definiert:
- die beteiligten Behörden und Stellen,
- die Art der Arzneimittelrisiken (z. B. Missbrauch, Fehlgebrauch, Nebenwirkungen, Gewöhnung, Abhängigkeit, Qualitätsmängel),
- die Verfahrensweise bei der Sammlung von Arzneimittelrisiken sowie
- das Vorgehen nach Gefahrenstufen:
- – Gefahrenstufe I: Informationsaustausch zwischen Bundesoberbehörde, Länderbehörden und betroffenem pharmazeutischen Unternehmer zur Ermittlung der Häufigkeit, des Schweregrades und der Ursachen;
- – Gefahrenstufe II: Anhörung des pharmazeutischen Unternehmers zu risikomindernden Maßnahmen, falls dieser nicht schon selbst solche Maßnahmen eingeleitet hat;
- der Maßnahmenkatalog, der von Stellungnahmen durch Sachverständige über Auflagen, Rückrufe bis zum Widerruf der Zulassung reicht.
Erkennung von Arzneimittelrisiken
Arzneimittelrisiken treffen die Apotheke auf zweierlei Weise:
- als unerwünschte Arzneimittelwirkung (UAW);
- als pharmazeutischer Qualitätsmangel des Arzneimittels.
In beiden Fällen besteht die Verpflichtung zur Meldung, zum einen schon aufgrund der Berufsordnungen der Kammern, zum anderen aufgrund des § 62 AMG (UAW) und des § 21 ApBetrO (pharmazeutische Qualitätsmängel).
Von der Risikoerfassung mithilfe des Stufenplans sind alle im Verkehr befindlichen Arzneimittel betroffen, auch jene, die einer Zulassung nicht bedürfen, beispielsweise weil sie aufgrund einer Standardzulassung nach § 36 AMG im Verkehr sind oder, wie alle Homöopathika, nur registriert sind.
Durch den Stufenplan wird gewissermaßen eine "Im-Markt-Dauerüberwachung" verwirklicht. Es ist daher für eine Apotheke, die eine UAW meldet, auch nicht sinnvoll, zwischen "bekannten" und "unbekannten" UAW unterscheiden zu wollen. In die Irre führt auch die Vorstellung, eine in der Gebrauchsinformation genannte UAW sei gewissermaßen schon nicht mehr interessant, daher auch nicht "meldewürdig". Oft sind UAW von Altwirkstoffen zwar bekannt, aber ihre Häufigkeit beruht auf einer unsicheren statistischen Grundlage. Das gilt vor allem auch bei Wirkstoffen im OTC-Bereich. Daher kann nur gelten:
Im Zweifelsfall immer melden!
Auch bei neuen Wirkstoffen kann bei der Markteinführung eine Aussage über UAW nur bedingt gemacht werden. Die untersuchten Patientenkollektive sind oft klein, sie können bei neuen Zytostatika beispielsweise zum Zeitpunkt der Zulassung nur wenige Hundert Patienten umfassen, jedoch führten Füllgraf und Palm zu Recht aus, dass bei einer UAW mit der Häufigkeit von 1:10.000 immerhin schon 40.000 Behandlungsfälle notwendig sind zur statistischen Sicherung mit 5% Irrtumswahrscheinlichkeit [4].
InternetPharmakovigilanzzentren |
Pharmakovigilanzzentren zur Erfassung von UAW
Die Erkennung von Arzneimittelrisiken beruht in Deutschland wesentlich auf dem Spontanmeldesystem, das weiterhin als unverzichtbar gilt, aber nur ein System der passiven Erfassung von UAW darstellt. Der Nachteil besteht in der häufig minderen Qualität der Berichte, sodass die Bewertung eines Zusammenhangs zwischen Arzneimitteleinnahme und eingetretener unerwünschter Arzneimittelwirkung sowie deren Häufigkeit nur schwer möglich ist. Deshalb wurden mit dem 12. Änderungsgesetz des AMG sogenannte Pharmakovigilanzzentren in das Überwachungssystem mit einbezogen; dort erfassen Ärzte bei Patienten, die zur stationären Behandlung aufgenommen werden, aktiv eventuelle UAW, indem sie sogenannte Triggersymptome verfolgen, und dokumentieren die UAW umfassend (Abb. 1).
Mängel der pharmazeutischen Qualität von Arzneimitteln
Die Verpflichtung, Qualitätsmängel, die vom Hersteller verursacht sind, unverzüglich der zuständigen Behörde zu melden (§ 21 Abs. 3 ApBetrO), sieht zunächst keine Beteiligung der AMK an diesem Meldeweg vor. Häufig besteht jedoch Unklarheit über den Verursacher, zumal bei jenen Arzneimitteln, die sich bereits im Verfügungsbereich des Patienten befanden oder bei denen eine Schadensentstehung auf dem Transportweg vermutet werden kann. Hier ist nicht unbedingt der pharmazeutische Unternehmer der Verursacher.
In vielen weiteren Fällen verfügt die meldende Apotheke auch nicht über die chemisch-analytischen Methoden, um einen Verdacht, beispielsweise auf Mindergehalt bei manipulierten Mehrdosenbehältnissen stark wirksamer Analgetika (s. u.), zu klären. Es bleibt der Apotheke deshalb unbenommen, einen entsprechenden Verdacht zunächst von der Arzneimittelkommission klären zu lassen.
Meldungen zu Medizinprodukten
Medizinprodukte entfalten ihre bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologisch oder immunologisch wirkende Mittel noch durch Metabolismus. Ihre Wirkung kann aber durch solche Mittel unterstützt werden, das heißt: Ein Medizinprodukt kann durchaus einen pharmakologisch wirksamen Arzneistoff enthalten.
Medizinprodukte tragen ein CE-Kennzeichen. Das Kennzeichen drückt aus, dass sie den sicherheitsrelevanten Normen entsprechen und in der gesamten EU verkehrsfähig sind. Sie tragen üblicherweise eine vierstellige CE-Kennnummer, die die "Benannte Stelle" kodiert, die die Zertifizierung durchgeführt hat. Wenn der Hersteller eine Selbstzertifizierung durchgeführt hat, was nur bei einfachen Medizinprodukten erlaubt ist, fehlt die Kennnummer.
Im Apothekenalltag können bei flüchtiger Betrachtung Medizinprodukte durchaus irrtümlich als Arzneimittel missverstanden werden. Beispielsweise wird manchmal beanstandet, dass die Zusammensetzung eines "Arzneimittels" nicht angegeben sei, bei genauerem Hinsehen handelt es sich dabei jedoch um ein Medizinprodukt. Bei Medizinprodukten ist aufgrund der Richtlinie 93/42/EG nur die nicht näher definierte Angabe "von Stoffen" gefordert, sodass beispielsweise die Angabe "mit Pflanzenextrakten" dieser Anforderung bereits genügt.
Meldungen zu Qualitätsmängeln bei Medizinprodukten sind nach § 3 Medizinprodukte-Sicherheitsplanverordnung außer an das zuständige BfArM auch an "die Kommissionen der Heilberufe" möglich, sofern "eine unverzügliche Weiterleitung an die Bundesoberbehörde" gewährleistet ist.
Bearbeitung von Risikofällen in der AMK
Zur Sicherstellung einer gleichbleibenden Bearbeitungsqualität hat die Geschäftsstelle der AMK ein Qualitätsmanagement-System eingeführt und arbeitet seit Juni 2008 nach DIN EN ISO 9001: 2000. Das Zertifikat wurde erteilt vom TÜV Hessen für die "Bearbeitung eingehender Meldungen aus Apotheken, Bewertung von Arzneimittelrisiken, Beauftragung des Zentrallabors, Informationen an Apotheken, Hersteller und Behörden, Bewertung von Rezepturen" (Abb. 2) [5]. Zur Bewertung der Risikoschwere verwendet die AMK-Geschäftsstelle die Klassifizierung des RAS (Rapid Alert System) der EMEA [6, 7]:
- Klasse I: Der vorliegende Mangel ist potenziell lebensbedrohend oder könnte schwere Gesundheitsschäden verursachen, z. B. wenn Deklaration und Inhalt nicht übereinstimmen, falsche Wirkstoffstärken mit schweren medizinischen Folgen vorliegen oder eine mikrobielle Kontamination von Iniectabilia oder Ophthalmica auftritt.
- Klasse II: Der vorliegende Mangel kann Krankheiten oder Fehlbehandlungen verursachen und fällt nicht unter Klasse I, z. B. fehlerhafte Kennzeichnung, falsche oder fehlende Produktinformation, Kontamination durch Fremdkörper oder chemische Verunreinigung, unzureichender Verschluss mit schweren medizinischen Folgen (bei Zytostatika oder fehlender Kindersicherung).
- Klasse III: Der vorliegende Mangel stellt kein signifikantes Risiko für die Gesundheit dar, z. B. falsche oder fehlende Angaben zu Charge oder Verfalldatum, Verschmutzungen, Abrieb.
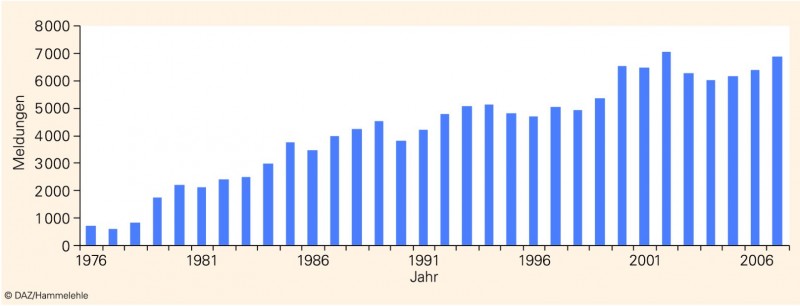
Meldevolumen kontinuierlich gestiegen
Dass die Apotheken alljährlich fast 7000 Arzneimittelrisiken an die AMK melden, ist keine Selbstverständlichkeit; dieses Meldevolumen wurde nach bescheidenen Anfängen erst nach jahrelanger Wechselwirkung mit der apothekerlichen Praxis erreicht (Abb. 3). Eine behördliche "Meldekontrolle" kann mithilfe des § 21 ApBetrO in der Praxis ohnehin nicht durchgesetzt werden. Das Erfolgsrezept für die kontinuierlich ansteigenden Meldezahlen beruht wesentlich auf
- der raschen Rückmeldung der AMK-Geschäftsstelle an die meldende Apotheke;
- der zügigen Information der meldenden Apotheke über das Ergebnis ihrer Meldung;
- der Integrität der AMK-Geschäftsstelle für die am Meldevorgang beteiligten Parteien;
- auf dem Warenersatz für die meldende Apotheke.
Durch die jahrzehntelange Tätigkeit der AMK-Geschäftsstelle liegt mittlerweile ein Datenbankbestand von fast 140.000 Meldungen vor, der eine rasche Suche nach ähnlichen Fällen erlaubt und damit eine bedeutsame Argumentationshilfe im täglichen Geschäftsverkehr mit pharmazeutischen Unternehmen, Behörden und Apotheken darstellt. Die Stellung der AMK-Geschäftsstelle als nachgeordnete, weisungsgebundene Abteilung eines marktbeteiligten, nicht eingetragenen Vereins (ABDA – Bundesvereinigung Deutscher Apothekerverbände) erlaubt die Offenlegung von qualitätsrelevanten Daten nur gegenüber den zuständigen Behörden und den betroffenen pharmazeutischen Unternehmen, nicht jedoch gegenüber einer breiten Öffentlichkeit, sodass die AMK auch kein Instrument für vordergründige PR sein kann. Die AMK-Geschäftsstelle verfügt naturgemäß über kein Initiativrecht.
Anlass für 20 bis 30 Rückrufe jährlich
Die Verantwortung für Rückrufe liegt immer in den Händen des betreffenden pharmazeutischen Unternehmers bzw. in der Verantwortung der zuständigen Behörde. Beide kommen üblicherweise zu einer gemeinsamen Einschätzung über die zu treffenden Maßnahmen der Risikoabwehr, sodass die zuständige Behörde nur noch die Durchführung der Maßnahmen des pharmazeutischen Unternehmers überwacht. Nur in den Fällen, in denen keine Einigung erreichbar ist, trifft die Behörde die nach § 69 AMG "zur Beseitigung festgestellter Verstöße und die zur Verhütung künftiger Verstöße notwendigen Anordnungen".
Es ist aber der Aufmerksamkeit der Apotheken zu verdanken, dass jährlich etwa 20 bis 30 Rückrufe aufgrund ihrer Meldungen an die AMK-Geschäftsstelle erfolgen. Dies belegt den Mehrwert an Arzneimittelsicherheit, der durch die Apotheken geschaffen wird. Umgekehrt gilt freilich auch: Nur eine ausreichend hohe permanente Meldetätigkeit über viele Jahre hinweg kann diesen Mehrwert sichern.
Aufschlüsselung der Meldungen
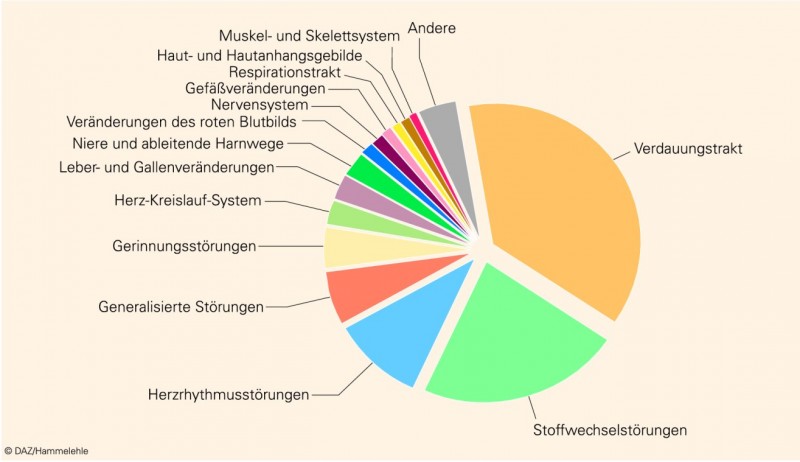
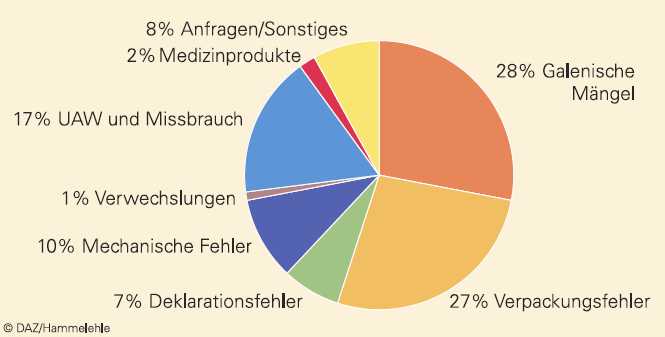
Seit vielen Jahren sind ungefähr drei Viertel der Meldungen pharmazeutischen Qualitätsmängeln zuzuordnen, während ein knappes Fünftel der Meldungen UAW betrifft (Abb. 4). Bei der Verteilung der pharmazeutischen Qualitätsmängel nehmen Beanstandungen zu galenischen Mängeln oder zur Verpackung den Hauptanteil ein, ein beachtlicher Teil der Beanstandungen betrifft jedoch auch mechanische Fehler, die meist bei Insulinpens oder Inhalatoren auftreten und in einer Vielzahl der Fälle nicht auf Fertigungsfehler, sondern auf Probleme bei der Anwendung zurückgeführt werden können. Medizinprodukte nehmen zwar im Zehn-Jahres-Durchschnitt nur einen geringen Anteil von 2% ein, sind jedoch im Zeitraum 2005 bis 2007 im Durchschnitt auf 3% gestiegen, was auch das Ausweichen von Arzneimittelherstellern in den Sektor der Medizinprodukte widerspiegeln mag.
Fallbeispiele aus der Praxis der AMK
Das Auftreten von Fremdkörpern in festen Arzneiformen ist zwar selten, kommt jedoch immer wieder einmal vor. Insgesamt verzeichnet die AMK-Datenbank z. B. 15 Fälle von in Tabletten verpressten Drahtstücken. Diese unmittelbar als RAS I einzustufenden Fälle mögen auch in Abhängigkeit der Bestückung von Produktionslinien mit Metalldetektoren nicht ganz zu vermeiden sein. Bisweilen lässt die Drahtform Rückschlüsse auf deren Herkunft zu (Abb. 5).
Eindrucksvolle Bilder der Fehlanwendung von Arzneimitteln liefern seit Jahren Inhalatoren. Hier ist nur allzu oft erkennbar, dass die Patienten mit der Anwendung überfordert sind und durch Fehlanwendung die Geräte zum Funktionsverlust bringen können. Insbesondere bei den Pulverinhalatoren führt das Ausatmen in das Gerät durch die feuchte Atemluft zum Verbacken des Pulvers im Ventilstift und den Bohrungen des Plastikaufsatzes (Abb. 6). Es sind gerade solche Bilder, die immer wieder auch die Notwendigkeit der Unterweisung der Patienten in der Apotheke unterstreichen.
Funktionsstörungen von Mehrdosenbehältnissen stark wirksamer Analgetika sollten immer den Verdacht auf eine mögliche Manipulation wachrufen [8]. Kratzer und Druckstellen an der Verbördelung sind hierfür genauso ein Indiz wie ein fehlendes Steigrohr, das in manchen Fällen schon bei der Wareneingangskontrolle durch die Apotheke entdeckt wurde, bevor das Arzneimittel in den Verfügungsbereich des Patienten gelangte (Abb. 7). Hier kann der gesamte Inhalt bereits durch Wasser ersetzt worden sein. Das fehlende Steigrohr soll die Apotheke oder den Patienten zur Retoure veranlassen und so den Verursacher verschleiern.
Ausblick
In den über 30 Jahren ihres Bestehens hat die AMK einen umfangreichen Datenbestand zu Arzneimittelrisiken in Apotheken aufgebaut. Dies war nur möglich durch das dauernde Engagement jener Apotheken, die ihrer Berufspflicht nachkamen und diese Risiken meldeten. Sie haben mit diesen Meldungen den Beitrag des Apothekerstandes für die Arzneimittelsicherheit erst möglich gemacht.
- Von der Klugheit des Berufsstandes wird es immer abhängen, dies zu bewahren,
- von seiner Weitsicht, es für die Zukunft zu sichern, und
- von seiner Vorsicht, sich nicht in parapharmazeutischen Trugbildern und Gaukeleien zu verfangen.
Literatur
[1] Trendelenburg P.: Grundlagen der allgemeinen und speziellen Arzneiverordnung. F.C.W. Vogel, Berlin 1929, S. 4 –5.
[2] Schröder J.M., Düppenbecker H., Müller-Oerlinghausen B., Scheler F.: Die Arzneimittelkommission der deutschen Ärzteschaft: von den Anfängen bis zur Gegenwart. Deutscher Ärzte Verlag, Köln 2003, S. 15–24.
[3] Schmidt G.: Zentrum Pharmakologie und Toxikologie, Medizinische Fakultät der Georg-August-Universität Göttingen, in: Philippu A.: Geschichte und Wirken der pharmakologischen, klinisch-pharmakologischen und toxikologischen Institute im deutschsprachigen Raum. Berenkamp, Innsbruck 2003, S. 245–247.
[4] Fülgraff G., Palm D.: Pharmakotherapie, Klinische Pharmakologie. Gustav Fischer Verlag, Stuttgart 1995, S. 4 –5.
[5] Zertifikat-Registrier-Nr. 73 100 2404; Audit-Bericht-Nr. 4183 4176; internes Audit: Qualitätsmanagement im Gesundheitswesen, Walter Pinkernell, Osnabrück.
[6] EMEA: Compilation of community procedures on inspections and exchange of information; www.emea.europa.eu/Inspections/docs/CoCP/CoCP_RapidAlertProc.
[7] Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten: Vorgehensweise bei Arzneimittelrisiken, Verbraucherbeschwerden und sonstigen Beanstandungen; www.zlg.de/download/AM/QS/12110103.pdf.
[8] AMK: Abhängigkeit von Schlaf- und Beruhigungsmitteln. Dtsch. Apoth. Ztg. 147 (30), 3352 (2007).
Anschriften der Verfasser:
Prof. Dr. Thomas Beck
Arzneimittelkommission der Deutschen Apotheker
Carl-Mannich-Str. 26,
65760 Eschborn
th.beck@abda.aponet.de
Prof. Dr. Volker Dinnendahl
Heddernheimer Landstr. 75,
60439 Frankfurt
v.dinnendahl@t-online.de



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.