


















- DAZ.online
- DAZ / AZ
- DAZ 30/2003
- S. Menzel, G. Geiß...
Pharmakologie
S. Menzel, G. GeißlingerBuprenorphin – Neue E
Chemische und pharmakologische Besonderheiten
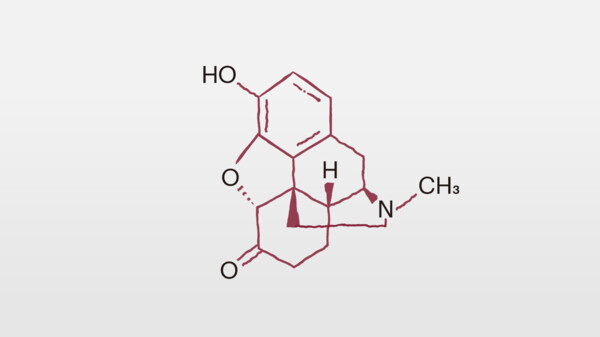
Buprenorphin ist ein Oripavin-Derivat, das halbsynthetisch aus Thebain hergestellt wird. Oripavin und Thebain sind die Hauptalkaloide des Armenischen Mohns (Papaver bracteatum), die sich von anderen Opioiden wie Morphin, Hydromorphon oder Levorphanol hauptsächlich durch die t-Butyl-Seitenkette am C7 unterscheiden (Abb. 1). Buprenorphin besitzt somit eine typische µ-Opioid-Struktur.
Die wichtigsten pharmakologischen Besonderheiten von Buprenorphin wurden bereits Ende der 70er-Jahre folgendermaßen beschrieben:
- Buprenorphin hat eine 25- bis 30-mal höhere Potenz als Morphin, aber abhängig von der verwendeten Spezies im Tiermodell eine niedrigere maximal erzielbare Wirkung (Ceiling-Effekt, s. u.).
- Dosis-Wirkungs-Kurven von Buprenorphin zeigen in manchen Tiermodellen einen außergewöhnlichen glockenförmigen Verlauf (die Wirkung nimmt bei Dosissteigerung ab einer bestimmten Dosis wieder ab).
- Die Wirkdauer von Buprenorphin ist vergleichsweise lang (nach sublingualer Applikation 6 bis 8 Stunden).
- Buprenorphin verursacht ein vergleichsweise mildes Abstinenzsyndrom und hat ein relativ geringes Abhängigkeitspotenzial.
- Für die Antagonisierung der Buprenorphin-Wirkungen sind im Tierversuch sowie beim Menschen relativ hohe Naloxon-Dosen erforderlich.
Wirkungsmechanismus und Wirkprofil
Interaktion mit Opioidrezeptoren Buprenorphin ist ein partieller Agonist am µ-Opioidrezeptor. Es besitzt einerseits eine hohe Affinität zum Rezeptor, andererseits aber eine niedrige intrinsische Aktivität [2 – 4]. Die intrinsische Aktivität ist ein Maß dafür, welche maximale Wirkung im jeweiligen biologischen System zu erreichen ist. Im Gegensatz zu Buprenorphin sind Morphin und Methadon in den meisten funktionellen Tests volle µ-Opioidrezeptor-Agonisten, sodass ihre intrinsische Aktivität größer ist als die von Buprenorphin.
Partielle Agonisten wirken im Gegensatz zu vollen Agonisten dualistisch: Bei Abwesenheit eines vollen Agonisten haben sie agonistische Eigenschaften; in Gegenwart eines vollen Agonisten schwächen sie dessen Wirkung ab und wirken somit antagonistisch [5]. Das Phänomen, dass ab einer bestimmten Dosis eine weitere Dosissteigerung keine Zunahme des Effektes bewirkt, wird als "Ceiling" bezeichnet und ist typisch für partielle Agonisten. Allerdings bezieht sich dieser Ceiling-Effekt nicht – wie häufig angenommen – nur auf den gewünschten therapeutischen Effekt, die Analgesie, sondern gleichermaßen auch auf die µ-Rezeptor-vermittelten unerwünschten Wirkungen [6 – 8].
Außer zu µ-Rezeptoren hat Buprenorphin auch eine hohe Affinität zu δ- und κ-Opioidrezeptoren, wobei die intrinsische Aktivität am κ-Rezeptor sehr gering ist (maximale Antwort 10%) und am δ-Rezeptor keine Wirkung ausgelöst wird. Am κ- und am δ-Rezeptor wirkt Buprenorphin daher also antagonistisch [9, 10]. Zum ORL1-Rezeptor hat Buprenorphin zwar eine niedrigere Affinität als zu den drei anderen Opioidrezeptoren, wirkt hier aber in Abhängigkeit vom Experiment entweder voll oder partiell agonistisch [11, 12]. Die Wirkungen, die durch Bindung an die verschiedenen Opioidrezeptoren ausgelöst werden können, sind in Abbildung 2 zusammengefasst.
Rezeptorkinetik Schon frühe Rezeptorbindungsstudien mit 3H-Buprenorphin konnten zeigen, dass sowohl die Assoziation mit dem Rezeptor als auch die Dissoziation vom Rezeptor langsam verlaufen [13, 14]. Verglichen mit Fentanyl, das auch eine sehr hohe Affinität zu Opioidrezeptoren besitzt, ist die Assoziation mit dem Rezeptor 3-mal langsamer, wobei die Halbwertszeit der Dissoziation mit 166 min mehr als 20fach länger ist.
Die Dissoziation von Fentanyl vom Rezeptor ist nach 1 Stunde vollständig, dagegen sind zum gleichen Zeitpunkt noch 50% der Bindungsstellen mit Buprenorphin besetzt. Während eine relativ geringe Naloxon-Konzentration von 2,5 nM genügt, um 50% Fentanyl-Moleküle von den Bindungsstellen zu verdrängen, ist für Buprenorphin eine 40fach höhere Naloxon-Konzentration erforderlich. Die besondere Rezeptorkinetik von Buprenorphin könnte für den lang anhaltenden analgetischen Effekt und die Notwendigkeit relativ hoher Naloxon-Dosen (5 – 10 mg) zum Antagonisieren der Buprenorphin-Wirkungen verantwortlich sein [15].
Dosis-Wirkungs-Beziehung im Tiermodell Die Dosis-Wirkungs-Kurven von Buprenorphin zeigen im Tierversuch in Abhängigkeit vom verwendeten Schmerzmodell entweder einen erwartungsgemäßen sigmoidalen oder aber einen außergewöhnlichen glockenförmigen Verlauf (Abb. 3). Bei der glockenförmigen Dosis-Wirkungs-Kurve nimmt die Wirkung ab einer bestimmten Dosis wieder ab.
Dies gilt sowohl für die erwünschte Analgesie als auch für unerwünschte Wirkungen wie Atemdepression und verzögerte Magen-Darm-Passage. Der maximale antinozizeptive sowie der maximale atemdepressive Effekt liegen im Tierversuch in Abhängigkeit vom verwendeten Testmodell zwischen 0,5 und 3 mg/kg Körpergewicht.
Bei gleichzeitiger Gabe eines Opioid-Antagonisten wie Naltrexon zeigt sich eine symmetrische Rechtsverschiebung der glockenförmigen Dosis-Wirkungs-Kurve (Abb. 3). Paradoxerweise sind dann sehr hohe Dosen von Buprenorphin, die allein kaum mehr antinozizeptiv wirken, in Gegenwart eines Antagonisten wieder sehr viel stärker wirksam [16].
Trotz intensiver Forschung kann man das Phänomen der glockenförmigen Dosis-Wirkungs-Kurven auch heute noch nicht vollständig erklären. Vor mehr als 20 Jahren haben Sadee et al. [17] die "nichtkompetitive Autoinhibitionstheorie" aufgegriffen und einen zweiten inhibitorischen Rezeptor postuliert, der in höheren Buprenorphin-Konzentrationen die Effekte des µ-Rezeptors aufhebt.
Als inhibitorischer Rezeptor wurde der κ-Rezeptor diskutiert. Möglicherweise trägt die agonistische Wirkung an ORL1-Rezeptoren, deren Aktivierung in manchen Schmerzmodellen Nozizeption bzw. Hyperalgesie bewirkt, zur glockenförmigen Dosis-Wirkungs-Kurve bei [12].
Dosis-Wirkungs-Beziehung bei Probanden Bei gesunden Probanden ist nach Gabe sehr hoher Buprenorphin-Dosen (> 8 mg, ein Vielfaches der mittleren Tagesdosis zur Schmerztherapie) gemäß seines partiellen Agonismus ein Ceiling-Effekt gegenüber subjektiv empfundenen Wirkungen ("How much do you feel the drug?") und auch gegenüber Miosis und Atemdepression beschrieben [7, 8] (Schmerzparameter wurden hier nicht untersucht). Die analgetischen Dosierungen sind allerdings niedriger und befinden sich auf dem linear ansteigenden Teil der Dosis-Wirkungs-Kurve, sodass eine Dosissteigerung auch eine stärkere Analgesie bewirkt. Somit behindert der Ceiling-Effekt die klinische analgetische Wirkung von Buprenorphin in der Regel nicht.
Von Vorteil kann der Ceiling-Effekt jedoch beispielsweise im Falle einer akuten Überdosierung von Buprenorphin sein. Dadurch, dass die Atemdepression nur bis zu einem maximalen Wert zunimmt und auch bei weiterer Dosissteigerung nicht mehr stärker wird, sondern eher abnimmt, ist das Risiko einer akuten Atemdepression verglichen mit reinen µ-Agonisten niedriger. Auch für das vergleichsweise geringe Missbrauchspotenzial von Buprenorphin wird neben der langsamen Rezeptorkinetik der partielle Agonismus verantwortlich gemacht [18, 19].
Pharmakokinetik
Resorption Buprenorphin ist eine außerordentlich lipophile Substanz (Octanol/Wasser-Verteilungskoeffizient 1943), die biologische Membranen nahezu ungehindert permeieren kann [20]. Nichtsdestoweniger erreicht nach peroraler Applikation nur ein sehr kleiner Teil der Dosis (ca. 15%) die systemische Zirkulation. Ursache dafür ist ein ausgeprägter First-pass-Effekt, d. h. die Substanz wird bereits bei der Passage der Magen-Darm-Wand und in der Leber in hohem Ausmaß metabolisiert.
Nach sublingualer Applikation ist die Bioverfügbarkeit unter Umgehung des First-pass-Effektes mit etwa 30 bis 50% sehr viel höher als nach peroraler Gabe, unterliegt aber aufgrund der Applikationsart sehr starken individuellen Schwankungen. Ähnliches gilt auch für die bukkale Anwendung. Auch nach transdermaler Applikation mithilfe eines Polymer-Matrixsystem-Pflasters (Transtec®) wird der First-pass-Effekt umgangen und Buprenorphin gut resorbiert [21]. Ziel des transdermalen therapeutischen Systems ist es, Buprenorphin über einen längeren Zeitraum konstant mit einer Kinetik 0. Ordnung freizusetzen. Im Steady State, der sich nach etwa drei Pflasterzyklen einstellt, werden dann relativ gleichmäßige, analgetisch wirksame Plasmaspiegel erreicht (vergleichbar mit Dauer-Infusion!).
Verteilung Buprenorphin ist zu 96% an Plasmaproteine der α - und β-Globulin-Fraktion gebunden. Dennoch verteilt es sich aufgrund seiner ausgeprägten Lipophilie sehr rasch, wobei zunächst hohe Konzentrationen in sehr stark durchbluteten Geweben wie Lunge, Herz, Niere, Leber und Gehirn gefunden werden, wie in Tierversuchen mit radioaktiv markierter Substanz gezeigt werden konnte. Dies bedingt den raschen Abfall der Plasmaspiegel unmittelbar nach Applikation der Substanz (Abb. 4). Die Buprenorphin-Konzentrationen im Gehirn übersteigen dabei die Plasmakonzentrationen.
Beim Rattenfötus konnte nachgewiesen werden, dass Buprenorphin die Plazentaschranke überquert. Entsprechende Ex-vivo-Experimente mit humanem Material weisen jedoch darauf hin, dass der Übertritt von Buprenorphin in den fötalen Kreislauf beim Menschen gering ist. Dies könnte auch das relativ seltene Auftreten von Entzugssymptomen bei Neugeborenen von opiatabhängigen Müttern, die während der Schwangerschaft mit Buprenorphin im Rahmen einer Substitutionstherapie behandelt wurden, erklären [22].
Metabolisierung und Elimination Buprenorphin wird nach oraler Gabe bereits im Gastrointestinaltrakt und bei der ersten Leberpassage (First-pass-Effekt!) sehr stark metabolisiert. Dabei wird die Substanz hauptsächlich mit Glucuronsäure zum Buprenorphin-3-glucuronid konjugiert. Außerdem wird Buprenorphin durch das Cytochrom-P450-Enzym CYP3A4 am Stickstoff oxidativ zu Norbuprenorphin desalkyliert (Abb. 5) [24], das seinerseits auch glucuronidiert wird. 50 bis 70% der Buprenorphin-Dosis werden via Fäzes ausgeschieden, 10 bis 30% findet man im Urin wieder, wobei in den Fäzes hauptsächlich nichtkonjugiertes Buprenorphin und Norbuprenorphin nachgewiesen werden, im Urin dagegen vor allem die konjugierten Verbindungen.
In welchem Ausmaß Norbuprenorphin gebildet wird, ist in der Literatur nicht eindeutig beschrieben. Es konnte allerdings gezeigt werden, dass bei der Substitutionsbehandlung von heroinabhängigen Patienten (Tagesdosis 8 mg Buprenorphin, sublingual) die Plasmaspiegel von Norbuprenorphin die der Muttersubstanz im Steady State übersteigen (0,8 ng/ml Buprenorphin versus 1,1 ng/ml Norbuprenorphin) [19].
Norbuprenorphin zeigt in Rezeptorbindungsstudien in vitro eine hohe Affinität zu µ-, δ- und κ-Rezeptoren und eine geringe Affinität zum ORL1-Rezeptor. In biochemischen Untersuchungen ist Norbuprenorphin ein partieller µ-Agonist, wirkt aber im Gegensatz zu Buprenorphin als voller Agonist am δ-Rezeptor. Am ORL1-Rezeptor ist Norbuprenorphin ein voller Agonist mit einer sehr niedrigen Potenz [4].
Aufgrund seiner größeren Polarität – der Octanol/Wasser-Verteilungskoeffizient von Norbuprenorphin ist 10-mal niedriger als der der Muttersubstanz – sind die Metaboliten-Konzentrationen im ZNS vergleichsweise niedrig, wie bei Ratten nachgewiesen werden konnte. Tierexperimente, die mit unterschiedlichen Schmerzmodellen durchgeführt wurden, liefern keine einheitlichen Ergebnisse zur antinozizeptiven Potenz von Norbuprenorphin. Während Norbuprenorphin im Tail-flick-Test bei der Ratte etwa 50-mal weniger antinozizeptiv wirksam ist als Buprenorphin [25, 26], ist seine antinozizeptive Potenz im Writhing-Test der Maus nur etwa 3-mal niedriger bei vergleichbarer maximaler Wirkung [4, 27].
Die für Buprenorphin in der Literatur beschriebenen Eliminationshalbwertszeiten divergieren. Während hauptsächlich ältere Arbeiten über Werte von ca. 5 Stunden berichten [20], geben jüngere Studien Werte von 25 bis 30 Stunden an [19, 30]. Aufgrund der niedrigen Buprenorphin-Dosis bewegen sich die Plasmaspiegel im Nano- bis Picogrammbereich je Milliliter.
Die Analysenmethoden waren in der Vergangenheit häufig nicht sensitiv genug, um die Buprenorphin-Plasmaspiegel über einen ausreichend langen Zeitraum zu quantifizieren, sodass zu kurze Eliminationshalbwertszeiten resultierten (Abb. 4). Ursache für die lange Eliminationshalbwertszeit von Buprenorphin ist eine enterohepatische Zirkulation der Substanz, wobei die biliär eliminierten Buprenorphin-Glucuronide im Darm gespalten und rückresorbiert werden können [28].
Klinisch-pharmakologische Besonderheiten
Anwendungsmöglichkeiten Buprenorphin (Temgesic®) ist bei starken und sehr starken Schmerzzuständen, die postoperativ, posttraumatisch, bei Herzinfarkt oder Tumoren auftreten, indiziert. Bei mittelstarken bis starken prolongierten nicht akuten Schmerzen eignet sich vor allem das Buprenorphin-Pflaster (Transtec®), das bei Durchbrechen von Schmerzspitzen mit sublingualem Buprenorphin und nach klinischen Erfahrungen mit schnell wirksamen Opioiden kombiniert werden kann [29, 30].
Eine Umstellung von Buprenorphin auf reine µ-Agonisten im Rahmen einer Opioidrotation und umgekehrt ist in der Regel ohne Probleme möglich. Eine Opioidrotation, d. h. ein Austausch eines stark wirksamen Opioids durch ein anderes stark wirksames Opioid, kann generell dann erforderlich werden, wenn die gewünschte Analgesie ausbleibt (z. B. wegen Toleranzentwicklung) oder wenn Nebenwirkungen wie Übelkeit oder Erbrechen die Schmerztherapie erschweren.
Ein relativ neues Einsatzgebiet von Buprenorphin in Deutschland, das in Frankreich schon etliche Jahre erfolgreich genutzt wird, stellt die Substitutionstherapie Opiat-Abhängiger dar. Buprenorphin ist nachgewiesenermaßen vergleichbar wirksam wie Methadon [31], mit dem Vorteil einer günstigeren Symptomatik nach Absetzen der Substanz: Entzugssymptome treten, bedingt durch die langsame Rezeptorkinetik von Buprenorphin und seine langsame Elimination, seltener und verzögert auf und sind in der Regel schwächer ausgeprägt als nach Methadon-Substitution [18].
Unerwünschte Wirkungen Das Nebenwirkungsprofil von Buprenorphin entspricht im therapeutischen Dosierungsbereich dem anderer µ-Opioide. Als unerwünschte Wirkungen werden Übelkeit, Erbrechen, Obstipation, Schwindel, Benommenheit, Kopfschmerzen, Sedierung, Dyspnö, Miktionsstörungen und Harnverhalten beobachtet. Nebenwirkungen wie Obstipation sind bei der Anwendung des Buprenorphin-Pflasters aufgrund sehr niedriger Wirkstoff-Spiegel im Gastrointestinaltrakt seltener als bei oral verabreichtem Morphin, und der Laxanzienbedarf ist dementsprechend niedriger [29].
Bei versehentlicher Überdosierung von Buprenorphin ist die Gefahr einer akuten Atemdepression aufgrund des Ceiling-Effektes geringer als bei reinen µ-Agonisten wie Morphin oder Methadon. Ab einer Dosis von 8 bis 32 mg nehmen die analgetischen Effekte und die Atemdepression nicht mehr zu. Sollte es dennoch zu einer Atemdepression kommen, sind relativ hohe Dosen von Naloxon (5 – 10 mg) zur Antagonisierung erforderlich, das eine wesentlich niedrigere Affinität zu µ-Rezeptoren besitzt als Buprenorphin [32]. Ferner ist die sehr viel kürzere Eliminationshalbwertszeit von Naloxon zu berücksichtigen [33].
Gelegentlich tritt unter langfristiger Opioidgabe eine Toleranzentwicklung auf, d. h., dass eine Dosissteigerung nötig ist, um den gleichen analgetischen Effekt zu erzielen [34]. Ursächlich dafür sind Adaptationsmechanismen an den Opioidrezeptoren (Desensibilisierung und ligandenbedingte Internalisierung) sowie ein durch das Opioid induzierter funktioneller Antagonismus. Es handelt sich dabei um intrazelluläre Aktivierungsprozesse, die wahrscheinlich über den NMDA (N-Methyl-D-aspartat)-Rezeptor vermittelt werden.
Im Tierversuch erfolgt die Toleranzentwicklung nach Opioidgabe sehr rasch und ausgeprägt. Bei Patienten mit chronischen Schmerzen ist sie dagegen vergleichsweise gering; eine Zunahme der Schmerzintensität ist bei ihnen sehr häufig nicht auf die nachlassende analgetische Wirkung, sondern auf eine Progredienz der Erkrankung zurückzuführen.
Vorteilhaft ist die "selektive Toleranzentwicklung" bezüglich Atemdepression, Übelkeit und Sedierung bei Patienten mit chronischen Schmerzen. Diese unerwünschten Effekte treten hauptsächlich bei Therapiebeginn auf und nehmen im Laufe der Behandlung ab. Keine Toleranz entwickelt sich jedoch gegen Obstipation und Miosis, die auch nach längerer Opioidgabe in gleichem Ausmaß nachweisbar sind.
Das Missbrauchspotenzial von Buprenorphin ist aufgrund des partiellen Agonismus und der langsamen Rezeptorkinetik geringer als bei reinen µ-Agonisten, da der Maximaleffekt auch bezüglich der euphorisierenden Wirkung reduziert ist (Ceiling!). Durch die vergleichsweise langsame Anflutung des Wirkstoffes und die gleichbleibenden Plasmaspiegel ist ein Missbrauch des buprenorphinhaltigen Matrixpflasters nahezu unmöglich.
Auch das Auftreten von Abstinenzsymptomen (Erregung, Angst, Nervosität, Schlaflosigkeit, Hyperkinesie, Zittern, Magen-Darm-Störungen), die durch eine opioidbedingte gegenregulatorische Zunahme der Adenylatcyclaseaktivität erklärt werden können, ist nach Langzeitanwendung von Buprenorphin vergleichsweise niedrig [18].
Anwendungseinschränkungen Bei Patienten mit Leberfunktionsstörungen kann die Intensität und Dauer der Buprenorphin-Wirkung aufgrund einer verzögerten Metabolisierung erhöht sein. Bei schwerer Leberinsuffizienz ist Buprenorphin daher kontraindiziert. Niereninsuffizienz dagegen gilt nicht als Kontraindikation. Wie alle Opioide darf Buprenorphin bei stark eingeschränkter Atemfunktion nicht angewendet werden.
Buprenorphin wird in Deutschland zur Therapie starker bis sehr starker Schmerzen sowie zur Substitutionstherapie Opiat-Abhängiger eingesetzt. Im Vergleich zu anderen arzneilich verwendeten Opioiden besitzt es ein einzigartiges Wirkprofil. Dazu zählen die Selektivität zu bestimmten Opioidrezeptoren, die langsame Rezeptorkinetik und der Ceiling-Effekt, der auch die euphorisierende Wirkung beschränkt und das Missbrauchspotenzial mindert.
Präparate
Buprenorphin steht für die Schmerztherapie als Injektionslösung (Temgesic®, i.v.; i.m.; 0,3 mg/ml), als Sublingualtablette (Temgesic®, 0,2 mg, 0,4 mg) und seit nahezu zwei Jahren als Pflaster (Transtec®, Freisetzungsraten: 35 µg/h, 52,5 µg/h, 70 µg/h; entsprechend einer Tagesdosis von 0,8, 1,2 und 1,6 mg) zur Verfügung.
Sublingualtabletten für die Substitutionstherapie Opiat-Abhängiger enthalten 0,4 mg, 2 mg oder 8 mg Buprenorphin (Subutex®).
Zusammenfassung
- Buprenorphin ist ein Opioid-Analgetikum mit einem einzigartigen Wirkprofil und einem großen Sicherheitsbereich.
- Buprenorphin hat eine hohe Affinität zum µ-Rezeptor, aber eine niedrige intrinsische Aktivität. Damit ist Buprenorphin ein partieller Agonist.
- Ein Ceiling-Effekt wird erst bei Dosierungen beobachtet, die die analgetischen Dosierungen deutlich übersteigen.
- Bei einer Überdosierung vermindert der Ceiling-Effekt das Risiko einer akuten Atemdepression.
- Eine durch Buprenorphin ausgelöste Atemdepression (selten!) kann wegen der hohen Rezeptoraffinität nur mit hohen Dosen Naloxon (5 – 10 mg) antagonisiert werden.
- Das Risiko einer physischen Abhängigkeit ist aufgrund der langsamen Rezeptorkinetik und Elimination von Buprenorphin vergleichsweise gering. Ein Abstinenzsyndrom nach Langzeitanwendung ist im Rahmen einer Schmerztherapie eher selten.
- Das Missbrauchspotenzial von Buprenorphin ist aufgrund des partiellen Agonismus kleiner als bei reinen µ-Agonisten.
- Aufgrund seiner langen Wirkdauer eignet sich Buprenorphin besonders zur Therapie mäßiger bis sehr starker chronischer Schmerzen, vor allem in Form des "Schmerzpflasters", da dieses eine konstante Wirkstoffmenge über einen längeren Zeitraum (3 Tage) freisetzt, bei Patienten eine hohe Akzeptanz findet und mit geringeren Nebenwirkungsraten behaftet ist.
- In höheren Dosierungen (sublingual) wird Buprenorphin erfolgreich zur Substitutionstherapie Opiat-Abhängiger eingesetzt.
- Die vollständige Aufklärung der molekularen Mechanismen des komplexen Wirkungsmusters von Buprenorphin wird ein spannendes Thema künftiger Forschungsarbeiten bleiben.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.