

















- DAZ.online
- DAZ / AZ
- DAZ 28/2021
- Stumpfe Antibiotika

Foto: asadykov/AdobeStock
Infektiologie
Stumpfe Antibiotika?
Von bedrohlichen Resistenzen, Innovationslücken und aktuellen Entwicklungen
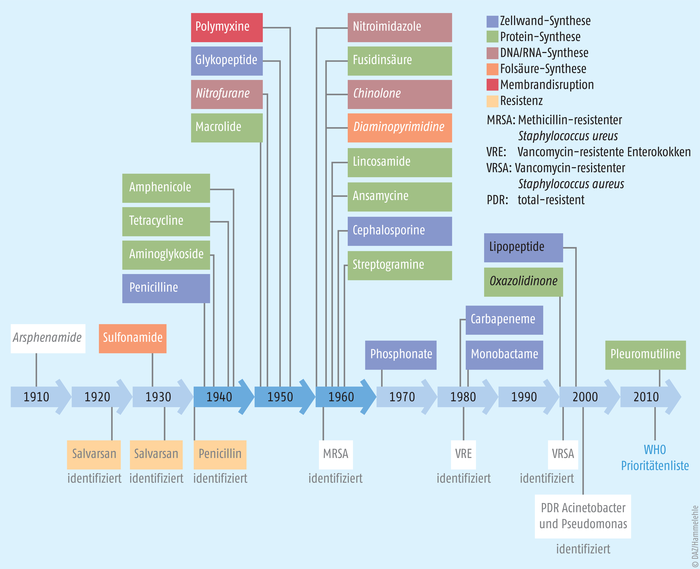
Antibiotikaresistenzen wurden schon mit der Einführung der ersten Wirkstoffe beobachtet (Abb. 1). Heute wissen wir, dass Resistenzfaktoren ganz natürlich zum genetischen Pool von Bakterien gehören und sogar in 30.000 Jahre alten Bakterien aus dem Permafrost nachgewiesen werden konnten [3]. Resistenzen können aber auch durch Mutationen spontan entstehen und mittels genetischer Austauschmechanismen auf andere Bakterien übertragen werden.
Abb. 1: Einführung neuer Antibiotikaklassen und Identifizierung einiger Resistenzen. Gezeigt werden systemische Applikationen, ohne Tuberkulose-Therapien. Synthetische Antibiotika sind kursiv, Wirkstoffe ohne heutigen therapeutischen Einsatz grau gekennzeichnet. 1940, 1950 und 1960 gelten als die „goldene Ära der Antibiotika“ (nach [Hutchings et al., 2019] und [Ventola, 2015]).
Der durch steigenden und oft unsachgemäßen Einsatz von Antibiotika entstehende Selektionsdruck führt zu resistenten Krankheitserregern. Als besonders gefährlich gelten Erreger, die gegen mehrere (multi drug resistant, MDR), sehr viele (extensively, XDR) oder sogar alle (pan, PDR) zugelassenen Antibiotika resistent sind [4]. Schätzungen gehen davon aus, dass ohne neue Antibiotika bis zum Jahr 2050 weltweit zehn Millionen Menschen pro Jahr an resistenten Keimen sterben könnten [5]. Ein reduzierter, sachgerechter Einsatz von Antibiotika kann zwar die Resistenzentwicklung verlangsamen, dieses Phänomen aber nicht verhindern. Daher ist die Entwicklung neuer, resistenzumgehender Antibiotika gegen neue Targets notwendiger denn je.
Innovationslücke in der Entwicklung
Obwohl Bakterien eine Vielzahl von potenziellen Angriffsorten aufweisen, besitzen zugelassene Antibiotika nur wenige generelle Wirkmechanismen [6]. Dazu gehören die Beeinflussung von Zellwand-, Protein-, DNA/RNA- und Folsäure-Biosynthese sowie der Membranfunktion [6]. In den letzten 20 Jahren wurden nur drei neuartige Antibiotikaklassen zugelassen: eine möglicherweise fatale Innovationslücke. Wie kam diese zustande? Die kurze Antwort wäre: Es ist sehr schwierig und teuer, neue Antibiotika zu finden, zu entwickeln und zuzulassen. Tatsächlich ist die Problematik deutlich komplexer. Es ist überaus schwierig neue Antibiotika gegen die problematischsten Erreger zu finden. Das sind vor allem Gram-negative Bakterien. Die Weltgesundheitsorganisation WHO hat dazu schon 2017 die entsprechenden Keime in einer Prioritätenliste (s. Tab. 1) zusammengefasst [7].
Priorität | Bakterium | Resistenz |
|---|---|---|
1: kritisch | Acinetobacter baumannii | Carbapenem |
Pseudomonas aeruginosa | Carbapenem | |
Enterobacteriaceae | Carbapenem + 3. Generation Cephalosporine | |
2: hoch | Enterococcus faecium | Vancomycin |
Staphylococcus aureus | Methicillin + Vancomycin | |
Helicobacter pylori | Clarithromycin | |
Camphylobacter | Fluorchinolone | |
Salmonella spp. | Fluorchinolone | |
Neisseria gonorrhoeae | 3. Generation Cephalosporine + Fluorchinolone | |
3: mittel | Streptococcus pneumoniae | Penicillin-unempfindlich |
Haemophilus influenzae | Ampicillin | |
Shigella spp. | Fluorchinolone |
Mikrobielle Naturstoffe sind eine herausragende Quelle für Antibiotika: 89% der von 1981 bis 2019 zugelassenen Antibiotika basieren auf Naturstoffen [8]. Obwohl das Potenzial der Naturstoffe längst nicht ausgeschöpft ist, stoßen Forscher immer wieder auf die gleichen Moleküle, denn die mit den erprobten Methoden identifizierbaren Stoffe sind oft schon bekannt [9]. Auch das Hochdurchsatz-Screening von synthetischen Stoffbibliotheken war bisher nicht erfolgreich. So führte GlaxoSmithKline eine Vielzahl an Hochdurchsatz-Screenings mit insgesamt einer halben Million Verbindungen durch. Daraus schaffte es nur eine Strukturklasse als Hit-Substanz in klinische Studien, da keine ausreichende Aktivität gegen Pathogene oder unspezifische Toxizität auftrat [6, 10]. Auch AstraZeneca blieb mit ähnlichem Aufwand erfolglos [11]. Die wenigen pharmazeutischen Firmen, die auf diesem Gebiet forschen, machten ähnliche Erfahrungen, die aber unpubliziert blieben. Dies zeigt eine weitere Schwierigkeit: Die Anforderungen an neue Antibiotika sind extrem hoch. Um die Penetration der Wirkstoffe in die bakteriellen Zellen zu gewährleisten, sind besondere physikochemische Eigenschaften der Moleküle notwendig. Vor allem die zusätzliche Zellmembran der Gram-negativen Keime ist eine Herausforderung [12]. Im Gegensatz zu den meisten in Hochdurchsatz-Screenings getesteten synthetischen Substanzen besitzen Naturstoffe hierzu oft die „richtigen“ physikochemischen Grundvoraussetzungen [11 – 13]. Die Wirkstoffe sollten zudem selektiv gegen pathogene Keime wirken, aber nicht toxisch für den Wirt und dessen gesundes Mikrobiom sein. Da in der Regel hohe Dosen des Antibiotikums vom Patienten toleriert werden müssen, ist eine große therapeutische Breite notwendig.
Auch die Anforderungen in den Zulassungsverfahren sind besonders hoch, da aus ethischen Gründen nicht gegen Placebo getestet, sondern eine Gleich- oder Mehrwertigkeit zur Standardmedikation erwartet wird. Dies ist wiederum schwierig, da glücklicherweise hochresistente Keime derzeit nicht der Normalfall sind. Während dieser Verfahren wird nicht die Aktivität gegen einen einzelnen Keim beurteilt, sondern der Therapieerfolg im Zusammenhang mit einer spezifischen Indikation, häufig hervorgerufen durch ein Spektrum an Pathogenen [4, 13]. Ein schwer zu erzielendes Maß an Selektivität ist also gefragt.
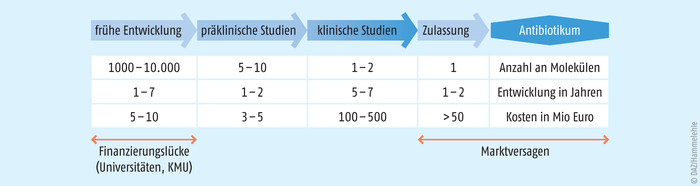
Die Entwicklung von Antibiotika ist teuer, da durch die im Vergleich zu anderen Wirkstoffen hohen Anforderungen und Schwierigkeiten die Forschung sehr kostenintensiv machen bei geringer Erfolgsrate. Hinzu kommt, dass Antibiotika ökonomisch wenig attraktiv sind, da durch ihren kurzen und möglichst sparsamen Einsatz die Aussicht auf Investitionsrentabilität kaum gegeben ist (Abb. 2).
Abb. 2: Weg eines Breitband-Antibiotikums durch die Pipeline mit Finanzierungslücken in der frühen Entwicklung durch akademische Institutionen und kleine bis mittelgroße Unternehmen (KMU). Die geringe Rentabilität führt zur Marktlücke (nach [Miethke 2021]).
Eine Therapie ist in der Regel wesentlich günstiger als bei den meisten medikamentös beeinflussbaren Krankheiten. Dies spiegelt sich letztlich in den Investitionen in die Antibiotikaforschung wider: Laut der Wirtschaftszeitschrift „brandeins“ liegt die jährliche Förderung der Antibiotikaforschung in der EU und den USA bei ca. 400 Millionen US-Dollar, während die jährlichen Forschungs- und Entwicklungskosten der zehn größten Pharmakonzerne sich auf 65 Milliarden US-Dollar summieren [15]. Die Innovationslücke in der Antibiotikapipeline ist also nicht nur auf technische Schwierigkeiten, sondern auch auf eine zu geringe (finanzielle) Wertschätzung und Entlohnung zurückzuführen. Dabei ist eine stetige, diverse Pipeline neuer Wirkstoffe erforderlich, um der Resistenzkrise entgegenzuwirken [14]. Eine sich entwickelnde Pandemie multi- und totalresistenter Keime würde die moderne Medizin in das präantibiotische Zeitalter zurückwerfen.
Wie schließt man die Innovationslücke?
Die Problematik antimikrobieller Resistenzen wird immer häufiger in Fachmedien und der allgemeinen Presse thematisiert. Eine Sensibilisierung unserer Gesellschaft für dieses Thema ist sehr wünschenswert, denn sie fördert durch mehr Wertschätzung den gezielteren Antibiotikaeinsatz und kann zu Förderprogrammen und besserer Entlohnung des Forschungsaufwandes führen – eine Grundvoraussetzung für die Entwicklung neuer Therapeutika [14]. Die WHO legte mit der Veröffentlichung der Erreger-Prioritäten-Liste erstmals den Grundstein für eine Anerkennung der Resistenzproblematik speziell durch Gram-negative Keime und forcierte so die Entwicklung von Antibiotika [7]. Vor allem „echte“ Innovationen werden dringend gebraucht, das heißt Antibiotika, die neue Zielstrukturen angreifen und mit neuen Wirkmechanismen keine Kreuzresistenzen mit Wirkstoffen aus der Klinik aufweisen [16, 17]. Bis es so weit ist müssen wir vor allem auf Wirkstoffe mit bekannten Struktur-Wirkungs-Beziehungen zählen. Die Optimierung bekannter Antibiotikaklassen hat den Vorteil, dass schneller das Aktivitäts- und Resistenzprofil sowie die unerwünschten Arzneimittelwirkungen verbessert werden können. Außerdem ist das unternehmerische Risiko geringer. Auf der anderen Seite sind die Optimierungsmöglichkeiten bei den gängigen Antibiotikaklassen fast ausgeschöpft und Kreuzresistenzen oft nicht auszuschließen. Die Optimierung ist demnach keine langfristige Lösung, könnte aber wertvolle Zeit im kontinuierlichen Kampf gegen antimikrobielle Resistenzen verschaffen. Auch die Tuberkulosetherapie ist aus globaler Sicht bedeutsam und mit einer äußerst großen Resistenzproblematik verbunden. Deshalb sind hier genauso Innovationen gefragt. Da die Anforderungen an die dort verwendeten Wirkstoffe den Umfang dieses Artikels übersteigen würden, wird die Tuberkulose-Problematik hier nicht intensiver aufgegriffen [18, 19].
Antibiotika in klinischer Prüfung
In den letzten 25 Jahren wurden lediglich drei neue Antibiotikaklassen zur systemischen Anwendung am Menschen zugelassen. Sowohl das synthetische Oxazolidinon Linezolid (2000) als auch die Naturstoff-basierten Antibiotika Daptomycin (zyklisches Lipopeptid, 2003) und Lefamulin (Pleuromutilin, 2019) wirken (fast) ausschließlich gegen Gram-positive Bakterien [13, 20].
Gegen Gram-negative Bakterien stehen mit der Zulassung von zwei neuartigen Klassen von β-Lactamase-Inhibitoren innovative Möglichkeiten für die Kombination mit Antibiotika zur Verfügung: Das 2015 zugelassene Diazabicyclooctan Avibactam und das zwei Jahre später eingeführte Boronat Vaborbactam reagieren kovalent reversibel mit der resistenzvermittelnden β-Lactamase. Avibactam und Vaborbactam zeichnen sich durch eine stärkere und breitere Wirksamkeit gegen viele β-Lactamasen und Carbapenemasen aus, als ältere β-Lactamase-Inhibitoren [13, 21]. Die fixen Kombinationen Avibactam/Ceftazidim und Vaborbactam/Meropenem werden bei komplizierten Harnwegsinfekten mit multiresistenten Gram-negativen Bakterien eingesetzt.
Angriffsort | Klasse | Name | Phase | Wirkspektrum | Besonderheit |
|---|---|---|---|---|---|
DNA/RNA | NBTI | Gepotidacin | III | G+, Ng | neues Target |
NBTI | Zoliflodacin | III | Ng | neues Target | |
Rifamycin-Chinolon-Hybrid | TNP-2092 | II | (G+) | ||
Rifamycin-Nitroimidazol-Konjugat | TNP-2198 | I | Ng | ||
Proteinsynthese | Makrolid | Solithromycin | ZB | G+ | |
Makrolid | Nafithromycin | II | G+ | ||
Oxazolidinon | Contezolid | ZB | G+ | ||
Aminoglykosid | EBL-10031 | I | (Ab), (Ent) | ||
Tetracyclin | KBP-7072 | I | Ab, G+ | ||
Tetracyclin | TP-271 | I | G+, (Ab) | ||
Tetracyclin | TP-6076 | I | Ab, (Ent) | ||
Metabolismus | FabI-Inhibitor | Afabicin | II | G+ | neuer Wirkmechanismus |
Zellteilung | FtsZ-Inhibitor | TXA709 | I | G+ | neuer Wirkmechanismus |
Zellhülle | Boronat-BLI+Cephalosporin | Taniborbactam + Cefepim | III | Ent, (Pa) | aktiv gegen Klasse A, B, C, D β-Lactamasen |
Boronat-BLI+Cephalosporin | VNRX-7145 + Ceftibuten | I | Ent | ||
Boronat-BLI+nicht deklariert | QPX7728 + QPX2014 | I | Ab, (Pa), Ent | ||
DBO-BLI/PBP2-Binder+β-Lactam-BLI | Durlobactam + Sulbactam | III | Ab | DBO-BLI antibiotisch wirksam | |
DBO-BLI/PBP2-Binder+Cephalosporin | Zidebactam + Cefepim | I | Ab, Pa, Ent | BLI antibiotisch wirksam, aktiv gegen Klasse A, B, C, (D) β-Lactamasen | |
DBO-BLI/PBP2-Binder+Carbapenem | Nacubactam + Meropenem | I | Ent, (Pa) | BLI antibiotisch wirksam, aktiv gegen Klasse A, B, C, (D) β-Lactamasen | |
DBO-BLI/PBP2-Binder+Cephalosporin | ETX-0282 + Cefpodoxim | I | Ent | BLI antibiotisch wirksam | |
DBO-BLI+β-Lactam | ARX-1796 | I | Ent | ||
Polymyxin | SPR-206 | I | Ab, Pa, Ent | ||
kationisches Peptid | PLG0206 | I | (Ab), (Pa), (Ent), G+ | neue chemische Klasse | |
nicht-traditionelle antibiotische Therapieansätze | |||||
Antikörper zur Bakterienbindung | 5 | I bis II | |||
Antikörper zur Toxinbindung | 3 | II bis III | |||
Bakteriophagen und -enzyme | 4 | I bis III | |||
Immunmodulatoren | 2 | I, ZB | |||
Toxin-Fänger | 1 | I | |||
Anti-Virulenzfaktoren | 3 | I bis II | |||
Eine weitere innovative Verbesserung in der Therapie gegen Gram-negative Bakterien war die Einführung von Cefiderocol, dem ersten Siderophor-Cephalosporin (2019). Durch seinen Eisen-komplexierenden Strukturteil kann der Wirkstoff über Eisen-Transporter via aktivem Transport in Bakterien eindringen. Zusätzlich ist Cefiderocol stabil gegenüber Metallo-β-Lactamasen und kann so gegen multiresistente Gram-negative Bakterien eingesetzt werden [18]. Stand September 2020 befinden sich 23 Antibiotika oder antibiotische Wirkstoffkombinationen in klinischen Studien, die von der WHO als „aktiv“ oder „möglicherweise aktiv“ gegen Pathogene der Prioritätenliste klassifiziert werden (s. Tab. 2) [22, 23]. Darunter sind fünf Vertreter aus vier neuen Antibiotikaklassen:
- Die beiden neuen bakteriellen Topoisomerase-Inhibitoren (NBTI) Gepotidacin und Zoliflodacin sind chemisch unterschiedlich und inhibieren die bakterielle Gyrase B an anderer Stelle als Fluorochinolone. Beide befinden sich in Phase III, sie wirken gegen Neisseria ghonorrhoeae und im Fall von Gepotidacin auch gegen Gram-positive Bakterien.
- In Phase II befindet sich Afabicin, das gegen Staphylococcus aureus wirkt, indem es ein Enzym der bakteriellen Fettsäuresynthese inhibiert.
- Die Substanz TXA-709 hemmt das Strukturprotein FtsZ, das in der Zellteilung eine wichtige Rolle spielt, und befindet sich in Phase I gegen multiresistente Staphylokokken (MRSA) [13, 18, 20].
- Das kationische Peptid PLG0206 wird in Phase-I-Studien gegen Infektionen mit S. aureus nach Gelenkstransplantationen getestet. Zur Zeit liegen wenig Informationen zu Wirkmechanismus, weiterem Spektrum, Toxizität und Stabilität vor [23].
Es wird vermutlich mindestens zehn weitere Jahre dauern, bis eine neue Wirkstoffklasse mit Aktivität gegen Gram-negative Bakterien auf den Markt kommt. Kurz vor diesem Punkt stand das makrozyklische Peptidomimetikum Murepavadin, das durch Bindung an das Lipoprotein LptD den Aufbau der äußeren Membran von Pseudomonas aeruginosa hemmt. Weil in den Phase-III-Studien vermehrt Nierenschäden auftraten, wurde die klinische Entwicklung für die systemische Anwendung beendet. Momentan wird die inhalative Anwendung von Murepavadin bei zystischer Fibrose in der Präklinik untersucht [20]. Die restlichen 18 Antibiotika und antibiotischen Wirkstoffkombinationen in den klinischen Studien sind zum Teil verbesserte Derivate bereits bekannter Antibiotikaklassen. Mit Aktivität gegen Gram-positive Keime zählen dazu
- die Makrolide Solithromycin (Zulassung beantragt) und Nafithromycin (Phase II),
- das Oxazolidinon Contezolid (Zulassung beantragt),
- das Tetracyclin TP-271 (Phase I) und
- der Rifamycin-Chinolon-Hybrid TNP-2092 (Phase II).
Auch gegen N. ghonorrhoeae befindet sich mit TNP-2198 (Phase I) ein Rifamycin-Nitroimidazol-Konjugat in der Entwicklung.
Einen Großteil der Wirkstoffkombinationen gegen Gram-negative Problemkeime bilden mit acht verschiedenen Vertretern die β-Lactamase-Inhibitoren in Kombination mit β-Lactamen. Besonders erwähnenswert ist hier die Kombination Taniborbactam-Cefepim (Phase III), da der Boronat-β-Lactamase-Inhibitor Taniborbactam gegen alle (bekannten) β-Lactamase-Klassen aktiv ist [20, 24]. Sehr interessant ist auch die Kombination von Cefepim mit dem Diazabicyclooctan-β-Lactamase-Inhibitor Zidebactam, die sich in Phase I befindet, sowie die Kombination von Nacubactam mit Meropenem (Phase I). Beide Kombinationen binden neben ihren Aktivitäten gegen fast alle β-Lactamase-Klassen an das Penicillin-Bindeprotein 2 (PBP-2) und besitzen eine intrinsische antibakterielle Aktivität. Noch einen Schritt weiter geht die Kombination Durlobactam und Sulbactam (Phase III), da hier der bekannte β-Lactamase-Inhibitor Sulbactam mit dem über PBP-2-Bindung antibakteriell wirksamen Diazabicyclooctan-β-Lactamase-Inhibitor Durlobactam kombiniert wird.
Ein Ausblick
- Der Blick in die klinische Pipeline ist ernüchternd: Keine neue Stoffklasse für Antibiotika mit Aktivitäten gegen Gram-negative Problemkeime ist in Sicht.
- Die frühe klinische Pipeline enthält einige innovative Projekte nicht-traditioneller Therapien, so dass hoffentlich neue Antibiotikaklassen und neue Therapieansätze es auf den Markt schaffen werden [17].
- Die präklinische Pipeline muss mit neuen und innovativen Antibiotika und anderen Ansätzen stetig „gefüttert“ werden, was nur durch Fortschritte in Grundlagen- und angewandter Forschung gelingen kann.
- Durch neue Erkenntnisse zu den Eigenschaften pathogener Bakterien sollten Screening-Methoden optimiert werden. Mit dem Wissen zu physikochemischen Eigenschaften von Molekülen, die in der Lage sind, Zellbarrieren Gram-negativer Bakterien zu überwinden, kann das Screening mit Hilfe der kombinatorischen Chemie optimiert werden [12, 13].
- Das bisher nur genetisch erfasste, aber bislang nicht in neuartige Naturstoff-Antibiotika übersetzte, biosynthetische Potenzial der Mikroorganismen sollte durch molekularbiologische, biotechnologische und bioinformatische Methoden in Zukunft weiter erschlossen werden [9, 13, 17].
- Eine bessere Finanzierung der Antibiotikaforschung ist notwendig. Wissenschaftlich existieren die Voraussetzungen für eine diverse und gut gefüllte Pipeline.
- Die Wertschätzung von Antibiotika bedarf allgemein größerer Unterstützung: Wenn Akzeptanz und Finanzierungsmodelle dazu führen, dass ein neu zugelassenes Antibiotikum nur als „Versicherung“ für selten auftretende Keime in der Schublade verbleibt, ist ein wichtiges Ziel erreicht [14].
Die einzigen nicht β-Lactame in klinischen Studien gegen Gram-negative Bakterien sind ein Aminoglykosid, zwei Tetracycline und ein Polymyxin (alle in Phase I) [13, 18, 20, 23]. Weiterhin nennt die WHO 27 nicht traditionelle, die Antibiotikatherapie unterstützende Therapieansätze in klinischen Studien. Die 18 systemisch angewandten Therapien umfassen acht Antikörper und Antikörperfragmente, vier Bakteriophagen-Mischungen oder -Endolysine, zwei Immunmodulatoren und vier weitere Ansätze [23]. Bei der Antikörpertherapie gibt es zwei Herangehensweisen:
- Entweder eine direkte Bindung an das Bakterium, die zur Opsonisierung und Phagozytose führt oder
- ein Abfangen bakterieller Toxine, um diese unschädlich zu machen.
Es sind fünf Antikörper gegen Staphylococus aureus, zwei gegen Pseudomonas aeruginosa und je ein Antikörperfragment gegen Campylobacter jejuni und Escherichia coli gerichtet [23]. Während Toxin-bindende Antikörper ihren Wert in der Klinik schon bewiesen haben, müssen bei opsonisierenden Antikörpern Fragen zu Wirkmechanismus und Wirksamkeit in klinischen Studien noch beantwortet werden [25].
Phagen-Endolysine sind Enzyme, die von Phagen zur Lyse von Bakterien produziert werden [13]. Zwei dieser Enzyme befinden sich in klinischen Studien gegen S. aureus. Außerdem befinden sich Gemische aus intakten Phagen in der Entwicklung patientenindividueller Zubereitungen gegen Escherichia coli und Klebsiella pneumoniae. Obwohl die Phagen-Therapie in Einzelfällen schon angewendet wird, ist es fraglich, ob sie mit den Zulassungsprozessen in der EU oder den USA vereinbar ist. Die Aktivität und Spezifität von Phagen ist unbestritten, ihnen stehen aber Probleme der schnellen Resistenzentwicklung, eines sehr engen Wirkspektrums und mögliche immunogene Reaktionen bei systemischer Therapie gegenüber [13, 23, 26].
Die beiden Immunmodulatoren (Reltecimod [Zulassung beantragt] und Rhu-pGSN [in Phase I/II]) regulieren die überschießende Immunantwort des Patienten auf bakterielle Toxine und können so den Infektionsverlauf positiv beeinflussen [13, 23]. Die vier weiteren Ansätze beinhalten ein Liposom alsToxin-Fänger und drei Anti-Virulenzfaktoren.
Bei der antivirulenten Therapie werden Substanzen eingesetzt, die die Schwere bakterieller Infektionen verringern, indem sie etwa in die bakterielle Kommunikation (Quorum Sensing) oder die Bildung von Virulenzfaktoren eingreifen. Als Anti-Virulenzfaktoren befinden sich momentan ein Sekretionssystem-, ein Adhäsions- und ein Biofilm-Inhibitor in klinischen Studien [13, 23, 26]. Die antivirulente Therapie ist ein interessanter und vielversprechender Ansatz, dessen praktische Eignung aber noch validiert werden muss.
Antibiotika in präklinischer Prüfung
Die den klinischen Phasen vorgeschaltete Präklinik umfasst Experimente zur Pharmakodynamik, Pharmakokinetik und Toxizität von Wirkstoffen in vitro und in vivo [27]. Im Gegensatz zu klinischen Studien gibt es zu präklinischen Projekten keine offiziellen Datenbanken, was eine realistische Einschätzung der Pipeline deutlich erschwert. Eine Auflistung aus dem Jahr 2020 nennt über 400 Projekte aus der antibakteriellen präklinischen Forschung [26]. Über 90% dieser oft innovativen Projekte sind in der Hand von Universitäten, Forschungseinrichtungen und kleinen bis mittelgroßen Unternehmen. Fast die Hälfte (46%) befassen sich mit Antibiotika und weitere 8% mit Wirkungsverstärkern wie β-Lactamase- oder Efflux-Pumpen-Inhibitoren. Ungefähr 70% der Antibiotika in der Präklinik haben andere Targets als bisher zugelassene Antibiotikaklassen und etwa die Hälfte zeigt Wirkung gegen Gram-negative Bakterien. Ein kleinerer Teil der Projekte (7%) befasst sich mit Antikörpern oder Antikörper-Antibiotika-Konjugaten. Den größten Anteil an Projekten für Behandlungsansätze, die bisher in westlichen Ländern nicht zugelassen sind, haben mit jeweils 8% die antivirulente Therapie und Therapien mit Phagen und Phagenbestandteilen [26]. |
Literatur
[1] Fleming A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to their Use in the Isolation of B. influenzæ. British journal of experimental pathology 1929;10:226–236
[2] Hutchings M, Truman A, Wilkinson B. Antibiotics: past, present and future. Curr. Opin. Microbiol. 2019;51:72–80, 10.1016/j.mib.2019.10.008
[3] D’Costa VM et al. Antibiotic resistance is ancient. Nature 2011;477:457–461, 10.1038/nature10388
[4] Ventola CL. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharmacy and Therapeutics 2015;40:277–283
[5] O’Neill J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. 2014
[6] Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 2007;6:29–40, 10.1038/nrd2201
[7] Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics - 2017. Informationen der Weltgesundheitsorganisation (WHO), www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf?ua=1
[8] Newman DJ, Cragg GM. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. Journal of natural products 2020; 10.1021/acs.jnatprod.9b01285
[9] Katz L, Baltz RH. Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol 2016;43:155–176, 10.1007/s10295-015-1723-5
[10] Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev 2011;24:71–109, 10.1128/CMR.00030-10
[11] Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov 2015;14:529–542, 10.1038/nrd4572
[12] Muñoz KA, Hergenrother PJ. Facilitating Compound Entry as a Means to Discover Antibiotics for Gram-Negative Bacteria. Accounts of chemical research 2021;54:1322–1333, 10.1021/acs.accounts.0c00895
[13] Lakemeyer M, Zhao W, Mandl FA, Hammann P, Sieber SA. Thinking Outside the Box-Novel Antibacterials To Tackle the Resistance Crisis. Angewandte Chemie (International ed. in English) 2018;57:14440–14475, 10.1002/anie.201804971
[14] Stern S et al. Breaking through the wall - A call for concerted action on antibiotics research and development. 2017
[15] Böttcher D. Die Antibiotika-Industrie in Zahlen. brand eins Wirtschaftsmagazin 2015:108
[16] Theuretzbacher U. Antibiotic innovation for future public health needs. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases 2017;23:713–717, 10.1016/j.cmi.2017.06.020
[17] Miethke M, Pieroni M, Weber T et al. Roadmap towards the sustainable discovery and development of new antibiotics. Nature Reviews Chemistry 2021; accepted
[18] Theuretzbacher U et al. Analysis of the clinical antibacterial and antituberculosis pipeline. The Lancet Infectious Diseases 2019;19:e40-e50, 10.1016/S1473-3099(18)30513-9
[19] Vjecha MJ, Tiberi S, Zumla A. Accelerating the development of therapeutic strategies for drug-resistant tuberculosis. Nature reviews. Drug discovery 2018;17:607–608, 10.1038/nrd.2018.28
[20] Butler MS, Paterson DL. Antibiotics in the clinical pipeline in October 2019. J Antibiot (Tokyo) 2020;73:329–364, 10.1038/s41429-020-0291-8
[21] Plescia J, Moitessier N. Design and discovery of boronic acid drugs. European journal of medicinal chemistry 2020;195:112270, 10.1016/j.ejmech.2020.112270
[22] Antibacterial products in clinical development for priority pathogens. Informationen der Weltgesundheitsorganisation (WHO), www.who.int/research-observatory/monitoring/processes/antibacterial_products/en/
[23] Antibacterial agents in clinical and preclinical development. an overview and analysis 2020. Informationen der Weltgesundheitsorganisation (WHO), www.who.int/publications/i/item/9789240021303
[24] Bill MK. Isolation and Characterization of New Antibacterial Active Secondary Metabolites from Microorganisms as Potential Starting Points for Drug Discovery. Dissertation. Justus-Liebig-Universität Gießen, 2021
[25] DiGiandomenico A, Sellman BR. Antibacterial monoclonal antibodies: the next generation? Curr. Opin. Microbiol 2015;27:78–85, 10.1016/j.mib.2015.07.014
[26] Theuretzbacher U, Outterson K, Engel A, Karlén A. The global preclinical antibacterial pipeline. Nat Rev Microbiol 2020;18:275–285, 10.1038/s41579-019-0288-0
[27] Blasius H. Arzneimittelentwicklung, präklinische und klinische Prüfung, Teil 1. DAZ 2014;22:56
Autoren
Joy Birkelbach studierte Pharmazie an der Universität Greifswald. Nach Diplomarbeit und Approbation arbeitete sie als Apothekerin in der Offizin. Im März 2019 begann sie ihre Promotion am Helmholtz-Institut für Pharmazeutische Forschung Saarland unter der Leitung von Rolf Müller und forscht an der Produktion und Strukturaufklärung von bioaktiven Naturstoffen.
Sebastian Walesch studierte Pharmazie an der Albert-Ludwigs-Universität Freiburg und schrieb seine Diplomarbeit an der Universität Cambridge. Nach Erhalt der Approbation arbeitete er als Apotheker in der Offizin. 2018 begann er seine Promotion in der Gruppe von Rolf Müller am Helmholtz-Institut für Pharmazeutische Forschung Saarland zur Produktion und Biosynthese antimikrobieller Naturstoffe aus Myxobakterien.
Peter Hammann studierte Chemie/Biochemie an der Technischen Universität Darmstadt, promovierte in organischer Chemie an der Leibniz Universität Hannover und habilitierte sich in medizinischer Chemie an der Veterinärmedizinischen Hochschule Hannover. Er war über 30 Jahre bei der Hoechst AG (heute Sanofi-Aventis) in Deutschland, Indien und USA vor allem für die Identifikation von Wirkstoffen im Bereich Antiinfektiva zuständig. Er ist Honorarprofessor seit 2016 an der Justus-Liebig-Universität Gießen und seit 2020 an der Universität des Saarlandes.
Foto: Oliver Dietze
Rolf Müller studierte Pharmazie an der Universität Bonn, wo er 1994 promovierte. Seit 2003 ist er Professor für Pharmazeutische Biotechnologie an der Universität des Saarlandes und seit 2009 Gründungsdirektor des Helmholtz-Instituts für Pharmazeutische Forschung Saarland. Rolf Müller koordiniert im Deutschen Zentrum für Infektionsforschung (DZIF) den Forschungsbereich „Neue Antibiotika“. Er wurde 2016 in die Nationale Akademie der Wissenschaften Leopoldina berufen und erhielt für seine Forschung 2003 den BioFuture Preis des BMBF und 2021 den Leibniz Preis der Deutschen Forschungsgemeinschaft.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.