




















- DAZ.online
- DAZ / AZ
- DAZ 48/2018
- Maßgeschneidert

Foto: kalou1927 – stock.adobe.com
Klinische Pharmazie
Maßgeschneidert
Die Pharmakogenetik ebnet den Weg für erfolgreiche individualisierte Interventionen
Auch heute sind in der Praxis noch viele Hürden auf dem Weg hin zu einer klinischen Anwendung von individualisierten Pharmakotherapien zu überwinden. Jedoch zeigt sich auch, dass wir der von Vogel beschriebenen Aussicht bereits einen großen Schritt näher gekommen sind. So konnte der klinische Nutzen der Anwendung einer individualisierten Pharmakotherapie nun in mehreren Studien in den USA gezeigt werden [2, 3].
Das richtige Arzneimittel, in der richtigen Dosis zur richtigen Zeit: Dies ist das Bestreben sowohl von Ärzten, Apothekern als auch Patienten und zudem das Ziel der individualisierten Pharmakotherapie. Die Individualisierung der Medizin ist deshalb seit jeher von großem Interesse. Beispielsweise sind schon aus dem 18. Jahrhundert Versuche bekannt, die Pharmakotherapie auf Patienten individuell zuzuschneiden. Der Arzt William Withering verwendete damals das Harnvolumen, um die Digitalis-Dosis für seine Patienten zu optimieren [4]. An eine Individualisierung auf Grundlage von DNA-Variationen war zu diesem Zeitpunkt aber noch lange nicht zu denken, geschweige denn die Existenz der DNA überhaupt bekannt. Mit der Entschlüsselung des humanen Genoms 2001 entstanden jedoch in den vergangenen Jahren vielversprechende Möglichkeiten: Die Kenntnis des menschlichen Genoms liefert die Chance, Methoden zu entwickeln, um Patienten nun individuell nach ihrem persönlichen Bedarf zu therapieren.
Wieso Individualisierung?
Randomisierte, kontrollierte klinische Studien gelten als Goldstandard für die evidenzbasierte Medizin, um eine wirksame und sichere Therapie zu gewährleisten. Allerdings gibt es zum Teil große interindividuelle Unterschiede bei Patienten, was zur Folge hat, dass manche Patienten nicht auf die Behandlung ansprechen, während andere unter Nebenwirkungen leiden. Grund dafür können demografische Faktoren wie Alter, Gewicht und BMI sowie Umweltfaktoren sein. Es gibt aber auch genetische Faktoren, die die Absorption, Verteilung und Metabolisierung von Arzneistoffen (Pharmakokinetik) sowie die Wirkung von Arzneistoffen auf den Körper (Pharmakodynamik) beeinträchtigen können. Pharmakogenomik und Pharmakogenetik sind entsprechende Fachgebiete, die sich mit dem Einfluss dieser genetischen Faktoren auf die Arzneimittelwirkung beschäftigen [5].
Pharmakogenomik und -genetik: was ist das?
Der Begriff der Pharmakogenetik wurde 1958 vom Humangenetiker Friedrich Vogel geprägt [1]. Er bezieht sich vor allem auf die klinische Praxis und beschreibt die interindividuellen genetischen Variationen, die Einfluss auf die Wirksamkeit und auf Nebenwirkungen von Arzneimitteln haben [6]. Dabei geht es besonders um die Gene, die den Metabolismus von Arzneistoffen und damit die Pharmakokinetik betreffen. Der später entstandene Begriff der Pharmakogenomik ist umfassender und beschäftigt sich mit allen Genen, die eine Variabilität von Arzneimittelwirkungen bei Patienten und damit primär eine Variabilität in der Pharmakodynamik zur Folge haben. Ziel der Pharmakogenetik und Pharmakogenomik ist es, eine patientenindividualisierte Pharmakotherapie zu ermöglichen [7, 8]. Durch ihre Anwendung ergibt sich die Chance, mittels Individualisierung der Pharmakotherapie sowohl die Wirksamkeit und Dosierung als auch die Sicherheit von vielen Arzneimitteln für viele Patienten zu optimieren.
CYP-Enzyme im Fokus
Mit der Entschlüsselung des humanen Genoms erwuchs auch die Möglichkeit, interindividuelle Differenzen im Genom von Patienten, sogenannte Polymorphismen, zu identifizieren und für die Arzneimitteltherapie nutzbar zu machen. Polymorphismen können sowohl die Effektgröße als auch die Plasmaspiegel von Arzneistoffen beeinflussen. Mithilfe von pharmakogenetischen Tests ist es nun allerdings möglich, den Einfluss von Polymorphismen auf die Arzneimitteltherapie vorherzusagen. Daraus resultiert die Aussicht, die Arzneimitteltherapie prospektiv durch die Auswahl geeigneter Medikamente und entsprechender Dosierungen zu optimieren. Dies hat im Gegensatz zum Therapeutic Drug Monitoring (TDM) den entscheidenden Vorteil, dass der Patient oder die Patientin nicht erst einer potenziell ineffektiven oder sogar toxischen Therapie ausgesetzt wird, bevor der Optimierungsprozess beginnen kann.
Ein Hauptaugenmerk wurde hierbei in den vergangenen Jahren auf Cytochrom-P450(CYP)-Enzyme gelegt. Es handelt sich um eine Superfamilie von Phase-I-Enzymen, die für den Abbau von körperfremden Substanzen, also auch Arzneistoffen, verantwortlich sind. Sie sind vorwiegend in der Leber, aber auch im Darm und in der Lunge zu finden [9]. Bis heute sind circa 60 verschiedene CYP-Enzyme beim Menschen bekannt. Ihre Nomenklatur ist durch die Aminosäuresequenz bestimmt. Genetische Polymorphismen führen zu einer Veränderung in der Aminosäuresequenz, was zu einer veränderten Enzymaktivität führen kann.
Diese unterschiedlichen Varianten werden dabei nach ihrer metabolischen Aktivität in vier Phänotypen unterteilt (siehe Tab. 1):
- Normale Metabolizer (NM; früher als Extensive Metabolizer (EM) bezeichnet),
- Intermediate Metabolizer (IM),
- Poor Metabolizer (PM) und
- Ultra-rapid Metabolizer (UM).
Phänotyp |
Eigenschaften der CYP-Enzyme |
Abkürzung |
|---|---|---|
Poor Metabolizer |
sehr geringe metabolische Aktivität
|
PM |
Intermediate Metabolizer |
erniedrigte metabolische Aktivität
|
IM |
Normal Metabolizer |
normale metabolische Aktivität
|
NM |
Ultra-rapid Metabolizer |
überschüssige Menge an funktionsfähigem CYP-Enzym
|
UM |
Bei PM und IM sind die hepatische und intestinale Metabolisierung von Arzneistoffen vermindert. Dies führt zu einer erhöhten Arzneistoffkonzentration im Blutkreislauf. Dadurch können vermehrt Nebenwirkungen auftreten, wenn der Arzneistoff zugleich ein enges therapeutisches Fenster aufweist. Im Gegensatz hierzu metabolisieren UM-Arzneistoffe durch eine erhöhte Anzahl von Enzymen schneller, sodass die Arzneistoffkonzentrationen im Körper geringer sind und der gewünschte Effekt entweder weniger oder gar nicht eintritt. Vorsicht ist allerdings geboten, wenn es sich bei dem Arzneistoff um ein Prodrug handelt, welches erst in einen aktiven Metaboliten umgewandelt werden muss. Ein Beispiel hierfür ist Clopidogrel (Plavix®). Clopidogrel ist ein Thrombozytenaggregationshemmer, der durch CYPs in einen aktiven Metaboliten umgewandelt wird. UM wandeln den Wirkstoff schneller und stärker um, während bei PM eine Wirkminderung zu beobachten ist. Die Fachinformation für Clopidogrel enthält daher einen Warnhinweis für PM, der besagt, dass PM einem erhöhten Thromboserisiko ausgesetzt sind.
CYP2D6, CYP2C9 und CYP2C19
Zum einen sind CYP-Enzyme an ca. 75% aller metabolischen Reaktionen im Körper beteiligt und nehmen somit eine entscheidende Rolle im Arzneistoffmetabolismus ein. Zum anderen können einige von ihnen eine große genetische Variabilität aufweisen. Daher ist eingehendes Verständnis dieser genetischen Variabilität und ihr Einfluss auf die Arzneimittelwirkung ein fundamentaler Bestandteil der Individualisierung der Arzneimitteltherapie, vor allem bei Arzneistoffen mit schmalem therapeutischem Fenster. CYP2D6 und 2C9 bzw. 2C19 sind circa zu je 20% an der Metabolisierung von Arzneistoffen beteiligt. Prominente Beispiele umfassen Codein, Tramadol (beide CYP2D6) und Phenprocoumon (CYP2C9). Aber auch Arzneistoffe in OTC-Präparaten wie Dextromethorphan (CYP2D6) und Omeprazol (CYP2C19) werden von diesen CYP-Enzymen verstoffwechselt (weitere Substrate siehe Tab. 2) [10, 11].
CYP-Enzym |
Substrate |
|---|---|
CYP2D6 |
Amitriptylin, Atomoxetin, Desipramin, Dextromethorphan, Imipramin, Metoprolol, Nebivolol, Perphenazin, Propranolol, Tramadol, Trimipramin, Venlafaxin, u. a. |
CYP2C19 |
Diazepam, Lansoprazol, Omeprazol, Rabeprazol, Voriconazol, u. a. |
CYP2C9 |
Celecoxib, Glimepirid, Phenytoin, Tolbutamid, Warfarin, u. a. |
Gerade bei diesen drei CYP-Enzymen tragen genetische Polymorphismen zu einer Veränderung der metabolischen Kapazität bei. Ca. 30% der Kaukasier sind beispielsweise CYP2C19-PM und -IM. Dies führt dazu, dass sich die Wirksamkeit und Verträglichkeit von CYP2C19-Substraten bei gleicher Dosis in zwei verschiedenen Patienten sehr stark unterscheiden können (interindividuelle Variabilität). Mithilfe von pharmakogenetischen Tests können die beschriebenen Enzym-Polymorphismen erkannt und die daraus gewonnenen Informationen über Arzneimittelwirkung und -metabolismus in die Therapie mit eingebaut werden.
Pharmakogenetische Testverfahren
Die Tests werden dabei meist nach dem Microarray-Verfahren durchgeführt. Soll beispielsweise der Genotyp eines Patienten für CYP2D6 ermittelt werden, wird eine Blutprobe oder ein Abstrich der Mundschleimhaut an ein qualifiziertes Labor geschickt. Die DNA wird dort zunächst isoliert, aufbereitet und ausgewählte, charakteristische Abschnitte mithilfe der Polymerasekettenreaktion (PCR) vervielfältigt [12]. Die mit einem Fluoreszenzmarker oder radioaktiv markierten PCR-Produkte werden anschließend auf einen kleinen Chip („Microarray“) aufgetragen, auf dem spezifische Oligonukleotide immobilisiert vorliegen. Beim Schritt der Hybridisierung paaren Abschnitte der amplifizierten DNA mit komplementären Oligonukleotiden. Ungepaarte DNA-Abschnitte werden anschließend abgewaschen. Die auf dem Chip gepaarten Abschnitte können nun photometrisch detektiert, die Signale mithilfe einer Software ausgelesen und mit einer Datenbank abgeglichen werden. In einer schriftlichen Testauswertung werden die Testergebnisse mit dem ermittelten Genotyp (EM, PM, IM oder UM) für CYP2D6 und Erläuterungen dem verordnenden Arzt mitgeteilt [12]. Mithilfe dieser Information kann dann die Auswahl von Arzneistoffen und Dosierungen auf den individuellen Patienten zugeschnitten werden. Darüber hinaus wäre eine Speicherung der Informationen in die für 2019 geplante elektronische Patientenakte eine Überlegung wert. Die Testergebnisse könnten so dem Arzt und Apotheker auch für zukünftige Individualisierungsfragen, in denen genetische Variabilität im Arzneistoffmetabolismus eine Rolle spielt, zur Verfügung gestellt werden. Somit würde der Patient auch weiterhin von den ermittelten Informationen profitieren.
Aktuelle Situation in Deutschland
Der Einsatz von pharmakogenomischen und pharmakogenetischen Tests in Deutschland ist im Gendiagnostikgesetz (GenDG) reguliert. Zwar zeigt sich die DPhG davon überzeugt, dass Apotheker „bestens qualifiziert sind, um arzneimittelbezogene Gentests zu veranlassen und die Ergebnisse gemeinsam mit dem Arzt und dem Patienten zu besprechen“ [13], jedoch sieht das GenDG vor, dass lediglich Ärzte eine „diagnostische genetische Untersuchung“ veranlassen und eine „genetische Beratung“ vornehmen dürfen [14]. Immerhin wird bereits heute bei 62 zugelassenen Wirkstoffen in der jeweiligen Fachinformation auf einen pharmakogenomischen Test hingewiesen. Bei über 80% davon, z. B. bei Imatinib oder Abacavir, handelt es sich sogar um einen Pflichttest, der vorgeschrieben ist, bevor das Arzneimittel bei einem Patienten angewendet werden darf [15]. Schwierigkeiten bei der Kostenübernahme durch Krankenkassen sind jedoch weiterhin ein Grund dafür, dass pharmakogenetische und pharmakogenomische Tests in der medizinischen Praxis in Deutschlandnoch nicht so stark etabliert sind wie beispielsweise in den USA.
Genotypisierung bei Brustkrebs
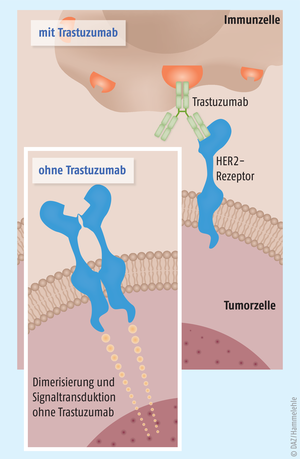
Das wohl prominenteste Beispiel für eine Genotypisierung in Hinblick auf die Pharmakodynamik ist die antitumorale Therapie des metastasierten HER2-positiven Mammakarzinoms mit Trastuzumab (Herceptin®). HER2 ist ein epidermaler Wachstumsfaktor-Rezeptor, der bei Aktivierung die Zellproliferation stimuliert und den programmierten Zelltod inhibiert. Daher weisen HER2-positive Tumore eine erhöhte Malignität auf. Bei ca. 20% der Brustkrebspatientinnen liegt sogar bereits im Frühstadium eine Überexpression des HER2-Rezeptors in den Tumorzellen vor, gegen den der monoklonale Antikörper Trastuzumab gerichtet ist. Patientinnen mit einer HER2-Überexpression im Tumorgewebe profitieren somit besonders von einer Therapie mit Trastuzumab, indem dieser die Dimerisierung des überexprimierten Rezeptors und damit die nachgeschaltete Signalkaskade blockiert und zusätzlich durch antikörpervermittelte zelluläre Toxizität Tumorzellen durch das Immunsystem gezielt eliminiert werden können (siehe Abb.). Die S3-Leitlinie zur Therapie des Mammakarzinoms sieht daher vor, dass Patientinnen vor Beginn der Therapie mithilfe eines pharmakogenomischen Tests genotypisiert werden und bei einer HER2-Überexpression die gezielte Therapie mit Trastuzumab begonnen wird [16, 17].
Praxis bestätigt Theorie
Dass die Pharmakogenetik bei der individualisierten Pharmakotherapie nicht nur theoretisch sondern auch praktisch ein enormes Potenzial mit sich bringt und Patienten helfen kann, zeigen nun die Ergebnisse von mehreren Studien aus den USA. Wissenschaftler um die Apothekerin Prof. Dr. Julie Johnson von der University of Florida konnten hierbei den großen Wert der individualisierten Therapie in der klinischen Praxis belegen. Die Arbeitsgruppe aus den USA ist Teil des IGNITE(Implementing GeNomics In pracTicE Network)-Projektes, dessen Ziel es ist, die Pharmakogenetik im klinischen Alltag zu implementieren. Dies soll gelingen, indem in klinischen Studien gezeigt wird, dass der Einsatz von pharmakogenetischen Tests eine Therapieoptimierung mit sich bringt. Patienten werden zunächst genotypisiert und den Ärzten anschließend das jeweils passende Medikament und die dazugehörige optimale Dosierung empfohlen.
Da sich in etwa die Hälfte der Leitlinien des Clinical Pharmacogenetics Implementation Consortium (CPIC®) mit Arzneistoffen befasst, die über die CYP-Enzyme CYP2D6 und CYP2C19 metabolisiert werden, stehen diese Therapien im Fokus der Wissenschaftler aus Florida. Eine davon ist die thrombozytenaggregationshemmende Therapie mit Clopidogrel. Clopidogrel ist – wie oben beschrieben – ein Prodrug, das vor allem über CYP2C19 bioaktiviert werden muss, bevor es wirken kann. Patienten, die Intermediate (IM) oder Poor Metabolizer (PM) für CYP2C19 sind, können den Arzneistoff somit kaum aktivieren. Dies ist bei 28% der kaukasischen, 36% der afroamerikanischen und sogar bei bis zu 70% der asiatischen Bevölkerung der Fall. Daraus resultiert für diese Patienten der Theorie nach ein erhöhtes Herzinfarkt- und Schlaganfallrisiko. Die Frage, die es nun jedoch noch zu klären gilt, ist, ob eine individualisierte Medizin auf Grundlage eines pharmakogenetischen Tests dieses Risiko reduzieren und somit die Implementierung der Pharmakogenetik in der Praxis unterstützen kann.
In der durchgeführten Studie wurde bei Patienten, die Clopidogrel erhielten, eine Genotypisierung für CYP2C19 empfohlen. Außerdem wurde gemäß der CPIC®-Leitlinie empfohlen, bei NM und UM die Standarddosis zu verabreichen, bei IM und PM (31,5% der Studienteilnehmer) hingegen eine alternative, CYP2C19-unabhängige Therapie (Prasugrel oder Ticagrelor) zu wählen. Die endgültige Entscheidung wurde aber jeweils vom behandelnden Arzt getroffen. Von den 572 Patienten, die IM oder PM waren, wurden circa 60% auf die empfohlene alternative Therapie eingestellt, circa 40% erhielten entgegen der Empfehlung trotzdem Clopidogrel. Die Ergebnisse hierbei sind deutlich: Das Risiko für ein schweres kardiovaskuläres Ereignis (in der Studie definiert als Herzinfarkt, Schlaganfall oder Tod innerhalb von zwölf Monaten) war im Vergleich zur alternativen Therapie fast dreimal so hoch für IM- und PM-Patienten, wenn sie trotz gegenteiliger Empfehlung Clopidogrel erhielten (23,4 Events pro 100 Patientenjahren, wenn der Empfehlung nicht gefolgt vs. 8,4 Events pro 100 Patientenjahren, wenn der Empfehlung gefolgt wurde). Hingegen bestand bei NM und UM mit Clopidogrel kein statistisch signifikanter Unterschied im Vergleich zur alternativen Therapie. Die Ergebnisse der Studie unterstützen somit eine Phänotyp- und damit Pharmakogenetik-basierte Therapie bei Clopidogrel [2].
Auch die Therapie mit dem oral verabreichten Antimykotikum Voriconazol, das ebenfalls über CYP2C19 metabolisiert wird, wurde von den Forschern aus den USA unter die Lupe genommen. Hierbei wirken sich niedrige subtherapeutische Plasmakonzentrationen negativ auf die klinische Wirksamkeit aus. Diese sind vor allem bei UM durch eine hohe Metabolisierungsrate zu erwarten. In einer klinischen Studie konnte nun gezeigt werden, dass mit ca. 52% aller UM deutlich mehr Patienten subtherapeutische Plasmakonzentrationen aufwiesen als die anderen Phänotypen (ca. 16%) [3]. Diese Unterdosierung wäre mit einer Genotyp-basierten Empfehlung vermeidbar. Weitere Studien in Bezug auf Empfehlungen für Protonenpumpenhemmer (PPIs), Opiate und Selektive-Serotonin-Wiederaufnahmehemmer (SSRI) laufen bereits und sollen ebenfalls den Mehrwert einer Pharmakogenetik-basierten individualisierten Therapie belegen.
Vielversprechende Studie auch in der EU
Auch in der EU läuft seit 2016 ein vielversprechendes Projekt, das die Implementierung von pharmakogenetischen Verfahren für die individualisierte Medizin vorantreiben will. Das Projekt, das unter dem Namen U-PGx bekannt ist, beschäftigt sich mit der Verbesserung der Arzneimittelsicherheit durch pharmakogenetische Typisierungen in Europa. Hierbei wird in sieben europäischen Ländern u. a. untersucht, ob die Genotypisierung von Patienten bei 43 verschiedenen Arzneimitteln wie Tramadol, Tamoxifen oder Atorvastatin zu besseren Behandlungsergebnissen führt. Damit soll sowohl Patienten geholfen, als auch ein Beitrag zu kostengünstigeren Therapien geliefert werden. Erste Ergebnisse der Studie, an der bereits über 3500 Patienten teilnehmen, werden für das Jahr 2020 erwartet [18, 19].
Somit ist man in einigen Fällen bereits in der Lage, auf Grundlage pharmakogenomischer und pharmakogenetischer Tests, die Therapie individualisiert zu gestalten. Die Identifizierung von Respondern, also solchen Patienten, die auf den Arzneistoff wie gewünscht ansprechen, und Non-Respondern ist bereits vor Beginn der Therapie möglich und muss nicht erst schmerzhaft während der Therapie festgestellt werden. Zudem kann die Häufigkeit von Nebenwirkungen durch besser eingestellte Wirkstoffspiegel gesenkt und damit die Arzneimitteltherapiesicherheit (AMTS) optimiert werden.
Das erwähnte Zitat von Vogel aus dem Jahr 1958, „Das gesamte Gebiet der Pharmakogenetik hat unseres Erachtens eine große Zukunft. […] So besteht Aussicht, dass wir dem Problem der großen biologischen Variationsbreite in der Reaktion auf Pharmaka, das die praktische Arzneimitteltherapie oft so stark hemmt, näher rücken werden“, hat somit auch heute noch Gültigkeit. Der genannten Aussicht sind wir aber durchaus bereits einen großen Schritt näher gekommen. |
Literatur
[1] Vogel F. Moderne Probleme der Humangenetik. Ergebnisse der Inneren Medizin und Kinderheilkunde 1959;12:52-125
[2] Cavalari L, Johnson JA et al. Multisite Investigation of Outcomes With Implementation of CYP2C19 Genotype-Guided Antiplatelet Therapy After Percutaneous Coronary Intervention. JACC Cardiovasc Interv 2018;11(2):181-191
[3] Johnson JA. Pharmacogenomics in Patient Care: The Future is Now. Vortrag in Münster 2017
[4] Somberg J, Greenfield D, Tepper D. Digitalis: 200 years in perspective. American Heart Journal 1986;3:615-620
[5] Högger P, Strehl E, Krämer I. Repititorium Klinische Pharmazie. 2015; 3. Auflage. Eschborn: Govi-Verlag
[6] Grimm T, Holinski-Feder E, Murken JD, Zerres K. Taschenlehrbuch Humangenetik. 2011; 8. Auflage. Stuttgart: Thieme
[7] Pirmohamed M. Pharmacogenetics and pharmacogenomics. Br J Clin Pharmacol 2002;52:345-347
[8] BfArM: Pharmakogenomik; www.bfarm.de/DE/Forschung/Pharmakogenomik/_node.html (zuletzt aufgerufen: 10.10.2018)
[9] Mutschler E, Geisslinger G, Menzel S, Ruth P, Schmidkto A. Pharmakologie kompakt, Allgemeine und Klinische Pharmakologie, Toxikologie. 2016; 1. Auflage, Stuttgart: Wiss. Verl.-Ges.
[10] Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & Therapeutics 2013;138(1):103-41
[11] FDA: Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. 2017; www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm (zuletzt aufgerufen: 10.10.2018)
[12] Schober D. Microassays, Genexpressionsanalyse und Bioinformatik. BIOspektrum 2002;3:307-310
[13] DPhG/RF. „Maßgeschneiderte Medikamente“ durch Gendiagnostik. 2012; www.aponet.de/aktuelles/ihr-apotheker-informiert/2012-10-massgeschneiderte-medikamente-durch-gendiagnostik.html (zuletzt aufgerufen: 10.10.2018)
[14] Gesetz über genetische Untersuchungen bei Menschen (Gendiagnostikgesetz – GenDG) vom 31. Juli 2009 (BGBl. I S. 2529, 3672)
[15] vfa. Die forschenden Pharmaunternehmen. In Deutschland zugelassene Arzneimittel für die Personalisierte Medizin. 2018; www.vfa.de/personalisiert (zuletzt aufgerufen: 10.10.2018)
[16] Baselga J. Treatment of HER2-overexpressing breast cancer. Annals of Oncology 2010;21:vii36-vii40
[17] Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF. Leitlinienprogramm Onkologie: Interdisziplinäre S3-Leitlinie für die Früherkennung, Diagnostik, Therapie und Nachsorge des Mammakarzinoms. Langversion 4.0. 2017
[18] U-PGx. Study Overview. www.upgx.eu/study (zuletzt aufgerufen: 10.10.2018)
[19] Cecchin E, Roncato R, Guchelaar HJ, Toffoli G. Ubiquitous Pharmacogenomics (U-PGx): The Time for Implementation is Now. An Horizon 2020 Program to Drive Pharmacogenomics into Clinical Practice. Curr Pharm Biotechnol 2017; 18(3):204-209
Autoren
Lukas Kovar hat an der Goethe-Universität Frankfurt/Main Pharmazie studiert. 2017/18 forschte er im Rahmen seines Praktischen Jahres an der University of Florida in den Bereichen der Pharmakogenetik und Pharmakokinetik in den Arbeitskreisen von Prof. Dr. Julie Johnson und Prof. Dr. Hartmut Derendorf.
Prof. Dr. Stephan Schmid ist Associate Professor an der University of Florida, Center for Pharmacometrics and Systems Pharmacology in Lake Nona (Orlando).
Prof. Dr. Hartmut Derendorf ist Distinguished Professor Emeritus des Departments of Pharmaceutics an der University of Florida in Gainesville, wo er seit 1983 Pharmakokinetik, Pharmakodynamik und Klinische Pharmakokinetik gelehrt hat.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.