














- DAZ.online
- DAZ / AZ
- DAZ 7/2016
- Mitochondrien als Ziel

Fotos: www.lhonsociety.org
Arzneimittel und Therapie
Mitochondrien als Ziel
Idebenon zur Therapie der Leberschen hereditären Optikusneuropathie (LHON) zugelassen
Elektronentransfer in der Mitochondrienmembran
In eukaryotischen Zellen sind die von einer Doppelmembran umgebenen Mitochondrien in vielerlei Hinsicht von zentraler Bedeutung. Neben der Einbindung in die Regulation der zellulären Calciumhomöostase und ihrer entscheidenden Rolle bei der Apoptose, liefern sie auch die nötige Energie für die Zelle. Zur Umwandlung der in Form von Glucose bzw. Fettsäuren gespeicherten Energie in das für die Zelle nutzbare Energiesubstrat Adenosin-5‘-triphosphat (ATP) verfügt das Mitochondrium über ein breites Spektrum an Enzymsystemen, die für die einzelnen Schritte der ATP-Synthese, aber auch für die Beseitigung gefährlicher Stoffwechselnebenprodukte essenziell wichtig sind.
Die Enzyme des Citratzyklus bzw. der β-Oxidation befinden sich in der Mitochondrienmatrix und setzen Glucose bzw. Fettsäuren zum aktivierten Metaboliten Acetyl-Coenzym A (Acetyl-CoA) um. Bei der weiteren Metabolisierung von Acetyl-CoA im Citratzyklus entstehen NADH und Succinat, die Ausgangssubstanzen der oxidativen Phosphorylierung (Oxphos) sind. Die oxidative Phosphorylierung wird durch die Komplexe I bis V der Atmungskette ermöglicht, die in die innere Mitochondrienmembran integriert sind. NADH und Succinat fungieren in den Komplexen I bzw. II als Donoren von Elektronen, die über Coenzym Q10 an den Komplex III und von dort über Cytochrom c an den Komplex IV weitergeleitet werden. In Komplex IV erfolgt schließlich die terminale Reduktion von Sauerstoff zu Wasser.
Während dieses Prozesses wird die beim Elektronentransfer freiwerdende Energie in einen elektrochemischen Gradient umgesetzt, indem Protonen von Komplex I, III und IV in den Raum zwischen äußerer und innerer Mitochondrienmembran gepumpt werden. Dieser Gradient dient letztlich im Komplex V als Treibstoff für die ATP-Synthese [1, 2].
Primäre mitochondriale Dysfunktion
Nach der gängigen Endosymbiontenhypothese sind Mitochondrien prokaryotischen Ursprungs und im Laufe der Evolution in die Eukaryotenzelle „eingewandert“. Dafür sprechen u. a. die Einfassung der Mitochondrien in eine Doppelmembran und das Vorhandensein eines eigenen Genoms, das in Form zirkulärer, doppelsträngiger DNA (mtDNA) vorliegt. In der mtDNA gibt es 37 Gene, welche für die Aufrechterhaltung der Oxphos notwendig sind, aber die überwiegende Mehrheit der im Mitochondrium vorhandenen Proteine wird von Genen im Zellkern kodiert, im Zytoplasma translatiert und schließlich in das Organell importiert [3, 4].
Im Gegensatz zur nukleären DNA erfolgt die Replikation der mtDNA stetig und unabhängig vom Zellzyklus, sodass in einem Mitochondrium mehrere Tausend Kopien der mtDNA vorliegen können. Tritt zu einem gegebenen Zeitpunkt eine Mutation in der mtDNA auf, kann diese Besonderheit dazu führen, dass neben der unveränderten mtDNA auch das mutierte Genom vervielfältigt wird, sodass letztlich ein Mitochondrium parallel über verschiedene Genome verfügen kann – ein Zustand, der als Heteroplasmie bezeichnet wird und u. a. für die hohe interindividuelle Variabilität in der Ausprägung mitochondrialer Erkrankungen verantwortlich ist.
Die Vererbung der mtDNA erfolgt rein maternal, da in der Zygote die väterlichen Mitochondrien abgebaut werden [5, 6]. Durch die Mutationen der mtDNA kann der konzertierte Ablauf der Atmungskette gestört werden, was zu gefährlichen Konsequenzen führt. Einerseits kann durch die kompromittierte Oxphos nur in eingeschränktem Maß ATP gebildet werden, sodass die Bereitstellung zellulärer Energie beeinträchtigt ist. Andererseits steigt jedoch auch die Konzentration reaktiver Sauerstoffspezies (ROS) im Mitochondrium an: Unter physiologischen Bedingungen werden 97 bis 99 Prozent des Sauerstoffs komplett zu Wasser reduziert. Nur ein bis drei Prozent werden nicht komplett reduziert, sodass das hochreaktive Superoxid (O2•–) entsteht. Dieses Radikal dismutiert spontan oder mittels Superoxiddismutase (SOD) zu Wasserstoffperoxid, das durch Glutathionperoxidase und Katalase zu Wasser abgebaut wird. Bei beeinträchtigter Effizienz der Atmungskette steigt der Anteil des inkomplett reduzierten Sauerstoffs an, sodass vermehrt ROS anfallen, die im schlimmsten Falle zu oxidativen Schäden an Nucleotiden, Lipiden und Thiolresten in Proteinen führen können [1].
Neuronen sind besonders anfällig für mitochondriale Dysfunktionen
Das zentrale Nervensystem ist in mehrfacher Hinsicht auf ein optimales Funktionieren der Mitochondrien angewiesen. Zum einen zeichnet sich das ZNS durch einen sehr hohen Energiebedarf aus – bei einem Anteil von nur zwei Prozent der Körpermasse verbraucht es ca. 20 Prozent der Gesamtenergie [7]. Des Weiteren verfügen Neuronen über keine ausgeprägten Glykogenreserven und eine reduzierte Glykolyseleistung, sodass sie auf eine stetige Versorgung mit Nährstoffen und Sauerstoff und eine gut funktionierende Oxphos angewiesen sind [8]. In den Membranen der Neuronen ist zudem der Anteil der mehrfach ungesättigten Lipide, die für oxidativen Stress besonders anfällig sind, relativ hoch. Bei ineffizienter Oxphos treten daher hier schnell Schäden auf.
Eine interessante pathophysiologische Querverbindung besteht auch zum Neurotransmitter Glutamat: Im Falle einer Energieverknappung, z. B. bei defektiver Oxphos, kommt es in Neuronen zu einer partiellen Depolarisation der Membran. Aus den sonst durch gebundenes Magnesium blockierten NMDA-Rezeptoren wird das zweiwertige Kation freigesetzt, und es kommt zur konstitutiven Aktivierung der NMDA-Rezeptoren, was die Neuronen schädigt (Exzitotoxizität) [9].
Der Sehvorgang und die Mitochondrien
Die Übermittlung der optischen Reize von der Netzhaut zum Gehirn wird durch die retinalen Ganglienzellen (RGC) gewährleistet, die nur teilweise myelinisiert sind. Die intraretinalen Anteile der RGC haben kein Myelin, was zur Folge hat, dass hier für die Signalübertragung viel mehr Energie benötigt wird, die nur durch eine sehr hohe Anzahl an Mitochondrien geliefert werden kann. So sind die RGC in ganz besonderem Maße anfällig für mitochondriale Dysfunktionen, und daher ist es nicht verwunderlich, dass viele Mitochondriopathien mit einer Verschlechterung des Sehvermögens einhergehen [10].
Lebersche hereditäre Optikusneuropathie (LHON)
Die prototypische und gleichzeitig häufigste mitochondrial verursachte Erkrankung des Nervus opticus ist die Lebersche hereditäre Optikusneuropathie (LHON). Es wird geschätzt, dass die Erkrankung in Europa nicht mehr als zwei von 100.000 Menschen betrifft, weshalb sie als seltene Erkrankung gilt. Die LHON betrifft typischerweise junge Männer im zweiten oder dritten Lebensjahrzehnt (80 – 90% der Patienten sind männlich). Sie ist klinisch gekennzeichnet durch eine einseitige subakute, schmerzlose Verschlechterung des Sehvermögens, auf die in einem zeitlichen Abstand von durchschnittlich sechs bis acht Wochen eine Verschlechterung des Sehens auch auf dem anderen Auge folgt. Dabei kommt es meist zur Ausbildung eines zentralen Gesichtsfeldausfalls, der sich mit der Zeit ausbreitet (s. Fotos am Anfang dieses Beitrags). Nach dieser durch rapiden Visusverlust geprägten Akutphase geht die Erkrankung in eine meist klinisch stabile chronische Phase über, während der es auch zu spontanen Besserungen des Sehvermögens kommen kann. Der Großteil der Patienten erfährt jedoch einen dauerhaften Verlust des Sehvermögens und gilt rechtlich gesehen als blind (Tab. 1) [11, 12].
Patientenkollektiv |
Haupterkrankungsalter: 20 – 40 Jahre
Geschlechtsprädilektion: 80 – 90% Männer
|
Prävalenz |
1/30.000 – 1/50.000 |
Symptome |
schmerzloser, rapider, zentral beginnender Sehverlust auf einem Auge; auf dem anderen Auge nach durchschnittlich 6 – 8 Wochen |
Ursachen |
Punkmutationen der mtDNA (Gene für Bestandteile des Komplex I der Atmungskette) |
Genetik |
rein maternal vererbt |
Prognose |
schlecht – die meisten Patienten beklagen eine dauerhafte Einschränkung des Sehvermögens, bleiben also sehbehindert |
Ursache der LHON sind Punktmutationen in der mtDNA, wobei 90 Prozent der Fälle durch eine von drei Mutationen an den Positionen 3460, 11.778 und 14.484 hervorgerufen wird. Die Punktmutation an Position 11.778 ist am häufigsten und weist auch die schlechteste Aussicht auf spontane Besserungen in der chronischen Phase auf [11]. Gemeinsam ist diesen Mutationen, dass durch sie eine Untereinheit des Komplex I der Atmungskette dysfunktional wird (Abb. 1) [13, 14]. Ob die daraus resultierende Überproduktion von ROS oder die eingeschränkte Bereitstellung von ATP oder beides für die Ausprägung der Symptomatik verantwortlich ist, ist bisher nicht geklärt [12].
Dass Männer bevorzugt von dieser Erkrankung betroffen sind, lässt sich nicht allein durch die Besonderheiten der mitochondrialen Vererbung erklären. Vielmehr werden ein X-chromosomal rezessiv vererbtes Suszeptibilitätsgen und hormonelle Faktoren als mögliche Ursachen dieser Geschlechtsprädilektion diskutiert [12].
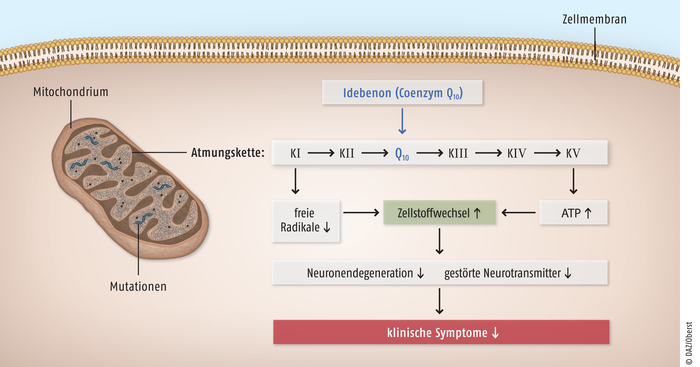
Abb. 1: Die Lebersche hereditäre Optikusneuropathie (LHON) ist eine Folge von Punktmutationen in der mitochondrialen DNA von Nervenzellen, die zu einer Dysfunktion im Komplex I der Atmungskette führt. Dadurch bildet das Mitochondrium weniger ATP und mehr freie Radikale, was beides auf die Dauer neurotoxisch wirkt. Idebenon (s. Abb. 2) steigert durch die direkte Übertragung von Elektronen auf den Komplex III der Atmungskette die ATP-Synthese und vermindert die Konzentration freier Radikale sowie die klinischen Symptome. [Quelle: Seifert R. Arzneimittelberatung bei seltenen Erkrankungen - Rare Disease Day Symposium, 20. Februar 2012, www.mh-hannover.de]
Therapie der LHON
Die klassische Therapie der LHON beruht auf rein supportiven Ansätzen wie der Bereitstellung von Sehhilfen, einer Rehabilitation für Sehbehinderte oder einem Coaching der Patienten, um Risikofaktoren wie Zigarettenrauch, Alkoholkonsum und mitochondrientoxische Medikamente zu identifizieren und zu minimieren [12].
Kausale Therapieansätze der LHON zielen bisher zum Großteil darauf ab, einen medikamentösen „Bypass“ für den dysfunktionalen Komplex I der Atmungskette zu schaffen und den oxidativen Stress zu beseitigen. Um den Komplex I der Atmungskette zu umgehen, wurde zunächst versucht, durch eine orale Therapie mit Ubichinon-10 (Coenzym Q10 , CoQ10) den Fluss der Elektronen durch die Atmungskette zu verbessern, allerdings ohne nennenswerten Erfolg. CoQ10 gehört zu einer Gruppe von Verbindungen, die durch ihren Chinonrest charakterisiert sind, sich aber in Länge und Struktur ihrer hydrophoben Seitenketten unterscheiden. Der Chinonrest ist für die biologische Funktion als Elektronenüberträger und Radikalfänger essenziell, nicht jedoch die Seitenketten, sodass CoQ10 -Analoga mit weniger hydrophoben Resten entwickelt werden konnten, die bei ähnlicher Wirkweise eine bessere Pharmakokinetik aufweisen [15].
Eines dieser Analoga ist das in Deutschland für die Behandlung der LHON zugelassene Idebenon (Abb. 2). Es weist am C10 eine OH-Gruppe auf und ist daher teilweise hydrophil, sodass es membranpermeabel ist – im Gegensatz zu CoQ10 , das membrangebunden ist. Während CoQ10 sowohl an Komplex I
als auch an Komplex II der Atmungskette als Elektronenüberträger dienen
kann, ist Idebenon lediglich in der Lage, Elektronen vom Komplex II der
Atmungskette zu Komplex III zu transferieren. Dagegen scheint Idebenon
am Komplex I mit CoQ10 um die Bindungsstelle zu
konkurrieren, ohne Elektronen für den Weitertransport aufzunehmen, wirkt
hier also als kompetitiver Inhibitor [15, 16].
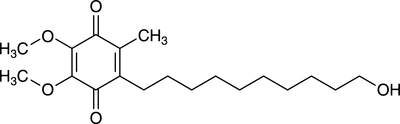
Abb. 2: Idebenon ist ein Derivat des Ubichinons (Coenzym Q10 , CoQ10) mit einer OH-Gruppe am C10 der Seitenkette. Dadurch ist es weniger hydrophob und kann die Zellmembran durchdringen. Im Zytosol wird es in Idebenol umgewandelt, indem die beiden Ketogruppen am Chinonrest zu Hydroxygruppen reduziert werden.
Da Idebenon durch seine geringere Hydrophobie auch im Zytoplasma gelöst vorliegt, kann es bereits nach dem Eintritt in die Zelle durch die zytosolische NADH-Chinon-Oxidoreduktase I (NQO1) in die biologisch aktive Hydrochinonform Idebenol umgewandelt werden. Idebenol wiederum kann in die Mitochondrienmembran diffundieren und dort durch den Komplex III der Atmungskette reoxidiert werden. Dies bedeutet, dass hier ein alternativer Stoffwechselweg genutzt wird, um Elektronen aus dem Zytosol in die Atmungskette zu schleusen. Aufgrund seiner Hydrophobie kann CoQ10dies nicht leisten. Ähnlich wie CoQ10 kann Idebenon in seiner Hydrochinonform als Radikalfänger dienen und so dem in einer dysfunktionalen Atmungskette entstehenden oxidativen Stress entgegenwirken.
Zusammenfassend lässt sich feststellen, dass Idebenon keinesfalls ein einfacher CoQ10-Ersatz ist, indem es freie Radikale abfängt. Vielmehr steigert es außerdem noch den Elektronenfluss der Atmungskette, indem vermehrt Elektronen aus dem Komplex II und zusätzlich aus dem Zytosol zum Komplex III geschleust werden (Komplex-I-Bypass) [15, 16].
Idebenon (Raxone®) hatte bereits im Jahr 2007 eine Zulassung als Orphan Drug erhalten. Am 8. September 2015 erhielt es die europaweite Zulassung für die Therapie der LHON „unter außergewöhnlichen Umständen“. Konkret bedeutet dies, dass die European Medicines Agency (EMA) sich vorbehält, jährlich eine Bewertung neuer Information vorzunehmen und den Zulassungsstatus gegebenenfalls anzupassen, denn aufgrund der Seltenheit der Erkrankung liegen erst weniger umfangreiche Informationen zu Nutzen und Risiko des Medikaments vor.
Zulassungsrelevant war die randomisierte, Placebo-kontrollierte, doppelblinde Phase-II-Studie RHODOS (Rescue of Hereditary Optic Disease Outpatient Study, SNT-II-003) mit 85 LHON-Patienten (SNT steht für den Hersteller Santhera Pharmaceuticals). Sie hatte ergeben, dass nach 24 Wochen ein Vorteil von Idebenon (900 mg/d) gegenüber Placebo bezüglich aller Endpunkte der Studie nachzuweisen war [17].
Eine anschließende Beobachtungsstudie (SNT-II-00-OFU), ein Expanded Access Program (SNT-EAP-001; einzelne LHON-Patienten erhielten auf Nachfrage Idebenon off label) und ein Case Record Survey (SNT-IR-006; Fallberichtsammlung von elf LHON-Behandlungszentren in Europa) bestätigten den Nutzen der Behandlung mit Idebenon [19].
Die häufigsten unerwünschten Wirkungen der Idebenon-Therapie sind
- Nasopharyngitis und Husten bei > 10 Prozent der Behandelten sowie
- Durchfälle und Rückenschmerzen bei 1 – 10 Prozent der Behandelten [18].
Obwohl sich der Nutzen der Behandlung aufgrund der geringen Patientenzahl schwer quantifizieren lässt, ist Idebenon der erste und bisher einzige Wirkstoff, welcher für die Behandlung einer mitochondrial bedingten Erkrankung zugelassen worden ist und eine konsistente Wirkung bei LHON aufweist. Der Wirkstoff birgt daher erhebliches therapeutisches Potenzial bei dieser seltenen, aber stark beeinträchtigenden Erkrankung. |
Literatur
[1] Szalardy L, et al. Electron Transport Disturbances and Neurodegeneration: From Albert Szent-Györgyi‘s Concept (Szeged) till Novel Approaches to Boost Mitochondrial Bioenergetics. Oxid Med Cell Longev 2015;2015:498401
[2] Smith RA, et al. Mitochondrial pharmacology. Trends Pharmacol Sci 2012;33(6):341-352
[3] Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet 2010;11:25-44
[4] Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol 2010;5:297-348
[5] Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol 2010;55(4):299-334
[6] Zeviani M, Di Donato S. Mitochondrial disorders. Brain 2004;127(Pt 10):2153-2172
[7] Papa S. Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications. Biochim Biophys Acta 1996;1276(2):87-105
[8] Almeida A, et al. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci USA 2001;98(26):15294-15299
[9] Zadori D, et al. Mitochondrial disturbances, excitotoxicity, neuroinflammation and kynurenines: novel therapeutic strategies for neurodegenerative disorders. J Neurol Sci 2012;322(1-2):187-191
[10] Chhetri J, Gueven N. Targeting mitochondrial function to protect against vision loss. Expert Opin Ther Targets, Epub 13.01.2016
[11] Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol 2005;140(3):517-523
[12] Meyerson C, Van Stavern G, McClelland C. Leber hereditary optic neuropathy: current perspectives. Clin Ophthalmol 2015;9:1165-1176
[13] Valentino ML, et al. The ND1 gene of complex I is a mutational hot spot for Leber‘s hereditary optic neuropathy. Ann Neurol 2004;56(5):631-641
(14] Chinnery PF, et al. The mitochondrial ND6 gene is a hot spot for mutations that cause Leber‘s hereditary optic neuropathy. Brain 2001;124(Pt 1):209-218
[15] Gueven N, Woolley K, Smith J. Border between natural product and drug: comparison of the related benzoquinones idebenone and coenzyme Q10. Redox Biol 2015;4:289-295.
[16] Jaber S, Polster BM. Idebenone and neuroprotection: antioxidant, pro-oxidant, or electron carrier? J Bioenerg Biomembr 2015;47(1-2):111-118
[17] Klopstock T, et al. A randomized placebo-controlled trial of idebenone in Leber‘s hereditary optic neuropathy. Brain 2011;134(Pt 9):2677-2686
[18] Santhera Pharmaceuticals (Deutschland) GmbH. Fachinformation zu Raxone, Stand 09/2015
[19] G-BA. Idebenon (Raxone®) Modul 4. Behandlung von Sehstörungen bei Jugendlichen und Erwachsenen mit LHON. Medizinischer Nutzen und medizinischer Zusatznutzen, Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen. Stand: 30.09.2015; www.g-ba.de/downloads/92-975-1157/2015-09-30_Modul4A_Idebenon.pdf
Autoren
Dr. med. Bastian Schirmer
Prof. Dr. med. Roland Seifert
Institut für Pharmakologie
Medizinische Hochschule Hannover
30623 Hannover



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.