

















- DAZ.online
- DAZ / AZ
- DAZ 16/2015
- Arzneimittel und ...
Wissenschaft
Arzneimittel und Transportproteine
Wie Transporter Invasion, Verteilung und Elimination regeln

Foto: lofik – Fotolia.com
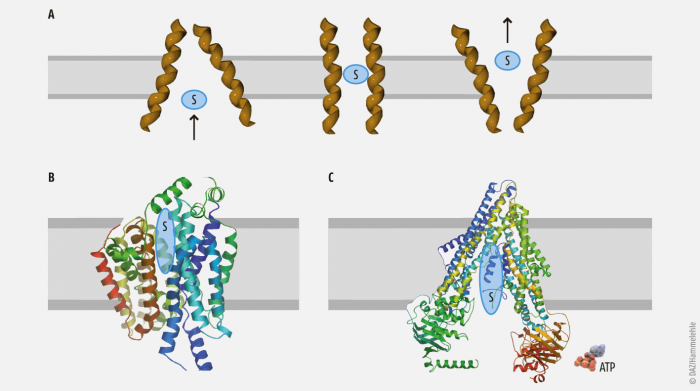
Häufig können Arzneistoffe aufgrund ihrer chemischen Eigenschaften wie Größe oder Ladung Biomembranen nicht oder nur sehr langsam überwinden. Transportproteine erleichtern den transmembranären Transport mithilfe eines Konzentrationsgradienten (sekundär aktiver Transport, z. B. SLC-Transporter) oder ermöglichen ihn unter ATP-Verbrauch (primär aktiver Transport, ABC-Transporter). Sie vermitteln teils die Aufnahme von Fremdstoffen oder körpereigenen Substanzen in die Zelle (Influx), teils deren Exkretion aus der Zelle (Efflux). Somit sind sie maßgeblich an den Phasen 0 und III des Arzneistoffmetabolismus beteiligt (Abb. 1).
C: P-gp (ABCB1) als Beispiel für primär aktive Transporter. Es bezieht die für den Transport notwendige Energie durch Dephosphorylierung von ATP.
Die über 400 bekannten Transportproteine werden in die Superfamilien der ABC-Transporter (ABC = ATP-binding cassette) und SLC (= solute carriers) unterteilt, die sich in ihrer Struktur, ihrem Substratspektrum und – häufig – in der physiologisch üblichen Richtung des Transports unterscheiden (Tab. 1):
- Die ABC-Transporter sind primär aktiv und sind vor allem für den Efflux verantwortlich.
- Die SLC sind sekundär aktiv und sind vor allem für den Influx verantwortlich.
| Genname | Andere Namen |
|---|---|
| SLC21 | SLC organic anion, SLCO; organic anion-transporting polypeptides, OATPs, s. Tab. 3 |
| SLC22 | organic anion/cation transporters, OATs/OCTs |
| SLC47 | multi-drug and toxin extrusion transporters, MATE |
| ABCB1 | multiple drug resistance protein 1, MDR1,permeability glycoprotein, P-gp, s. Tab. 2 |
| ABCC1 | multidrug resistance-associated protein 1, MRP1* |
| ABCC2 | multidrug resistance-associated protein 2, MRP2* |
| ABCG2 | breast cancer resistance protein 1, BCRP1* |
* vor allem relevant für Chemotherapeutika | |
Die Transportvorgänge Efflux und Influx beziehen sich immer auf eine Zelle. Sie können gegensätzlich sein, laufen aber meistens koordiniert ab. So können Influx und Efflux in Enterozyten eine Resorption aus dem Darmlumen ins Blut, in Hepatozyten hingegen eine Elimination aus dem Blut in die Galle bewirken.
Phase III des Metabolismus – Efflux
Der letzte Schritt des Arzneistoffmetabolismus (Phase III) ist die überwiegend biliäre oder renale Ausscheidung (s. Abb. 2). Die für den Efflux zuständigen ABC-Transporter kommen aber nicht nur in Leber und Niere vor, sondern auch in anderen Organen, wo sie den Körper vor Xenobiotika schützen (bes. Gastrointestinaltrakt, Blut-Hirn-Schranke, Plazenta, Hoden). In den Enterozyten tragen sie zum First-pass-Effekt bei, indem sie das Ausmaß der Bioverfügbarkeit eines Arzneistoffs beeinflussen. Auch in verschiedenen Knochenmark- und Blutzellen (Lymphozyten) kommt diese Membranpumpe vor, vermutlich ebenso zum Schutz vor toxischen Fremdstoffen.
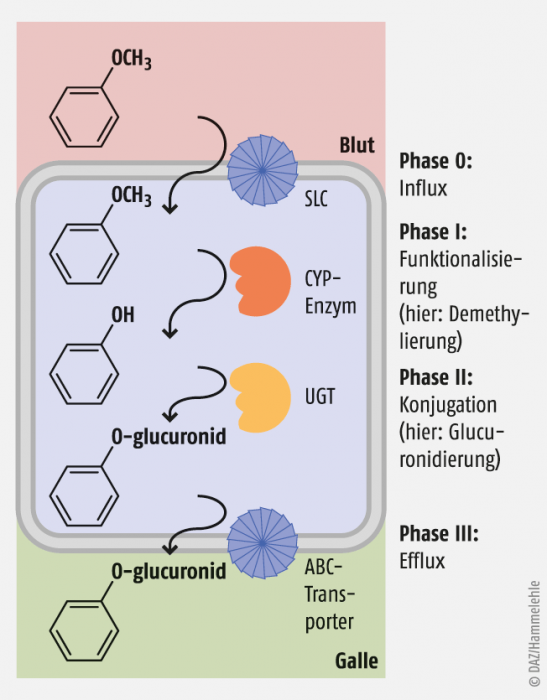
Abb. 2: Pharmakokinetische Phasen eines Wirkstoffs in einer Leberzelle (Hepatozyt). In diesem Beispiel wird Anisol SLC-vermittelt aus dem Blut aufgenommen, in der Zelle CYP-vermittelt funktionalisiert (demethyliert) und UGT-vermittelt glucuronidiert, dann schließlich mittels eines ABC-Transporters in die Gallenflüssigkeit ausgeschieden.
Die Gene, die die einzelnen ABC-Transporter kodieren, werden mit einem Großbuchstaben und einer Zahl bezeichnet, z. B. kodiert ABCB1 das „permeability glycoprotein“ P-gp (Tab. 1). ABC-Transporter wurden zuerst als Resistenzfaktoren von Chemotherapeutika (Zytostatika) erkannt und haben häufig entsprechende Synonyme wie MDR1 (multiple drug resistance protein 1). Da Transporter häufig auf der apikalen (nach außen gerichteten) Seite der Zellmembran exprimiert werden, wurden ihnen auch CD-Nummern zugeordnet, z. B. CD243 für P-gp (CD = cluster of differentiation, bezeichnet Oberflächenproteine von Zellen). Arzneistoffe, die in eine Tumorzelle gelangt sind, können von Transportern wieder aus der Zelle ausgeschleust werden, was als Chemoresistenz des Tumors bezeichnet wird.
P-Glykoprotein ist schon seit den 1970er Jahren bekannt und gut charakterisiert. Sein Spektrum von Substraten, Inhibitoren und Induktoren entspricht weitgehend demjenigen von CYP3A4 (Tab. 2; vgl. DAZ 2012, Nr. 40, S. 58 – 67). Die Substrate sind meist lipophile oder amphiphile Verbindungen mit einer Masse von 200 bis etwa 2000 Da. Neben vielen Arzneistoffen und Umweltgiften werden auch Nahrungsbestandteile und endogene Substanzen wie Hormone (Corticosteroide), Zytokine, Aminosäuren, Zucker und Peptide von P-gp transportiert.
P-gp-Substrate | |
| Antibiotika: | Erythromycin, Tetracyclin, Levofloxacin, Ofloxacin |
| Antidepressiva: | Amitriptylin, Nortriptylin, Doxepin, Venlafaxin, Paroxetin |
| Antiemetika: | Ondansetron |
| Antihistaminika: | Fexofenadin, Ranitidin, Terfenadin, Cimetidin |
| Antikoagulanzien: | Dabigatran, Apixaban, Rivaroxaban |
| Antikonvulsiva: | Phenytoin, Carbamazepin, Lamotrigin, Phenobarbital, Felbamat, Gabapentin, Topiramat |
| Antipsychotika: | Olanzapin, Amisulprid |
| Betablocker: | Carvedilol, Celiprolol, Talinolol |
| Ca2+ -Kanal-Blocker: | Verapamil, Diltiazem, Nifedipin, Nitrendipin, Felodipin |
| Chemotherapeutika: | Taxane, Vinca-Alkaloide, Anthracycline, Imatinib, Lonafarnib, Doxorubicin, Daunorubicin, Idarubicin, Epirubicin, Vinblastin, Vincristin, Etoposid, Teniposid |
| Herzglykoside: | Digoxin, Digitoxin |
| Immunsuppressiva: | Ciclosporin A, Tacrolimus, Sirolimus |
| Opioide: | Morphin, Methadon, Loperamid, Fentanyl |
| Proteaseinhibitoren: | Amprenavir, Indinavir, Ritonavir, Saquinavir, Nelfinavir, Lopinavir |
| Statine: | Atorvastatin, Lovastatin |
| Steroide: | Budesonid, Aldosteron, Dexamethason, Hydrocortison, Cortisol, Prednisolon, Methylprednisolon |
P-gp-Inhibitoren | |
| Antiarrhythmika: | Amiodaron, Chinidin, Chinin, Propafenon |
| Antibiotika: | Makrolide: Erythromycin, Clarithromycin |
| Antimykotika: | Itraconazol, Ketoconazol |
| Ca2+ -Kanal-Blocker: | Diltiazem, Felodipin, Nicardipin, Nifedipin, besonders Verapamil (nicht: Azithromycin) |
| Immunsuppressiva: | Ciclosporin |
| Proteaseinhibitoren: | Indinavir, Nelfinavir, besonders Ritonavir, Saquinavir |
P-gp-Induktoren | |
| Antidepressiva: | Hyperforin (Johanniskraut) |
| Antikonvulsiva: | Carbamazepin (weniger: Oxcarbazepin), Phenobarbital, Phenytoin, Primidon |
| HIV-Therapeutika: | Efavirenz |
| Tuberkulostatika: | Rifampicin |
| Zytostatika: | Enzalutamid |
Arzneistoffe können die Genexpression von P-gp induzieren oder seine Aktivität inhibieren. Die Induktion von P-gp wird hauptsächlich über den Pregnan-X-Rezeptor (PXR) vermittelt, der auch die Expression von CYP3A4 steigert. Durch Entzündungen kann es zur Herabregulation von P-gp kommen. Eine veränderte Funktion bzw. Expression von P-gp hat einen großen Einfluss auf die Behandlung neurologischer oder psychiatrischer Erkrankungen. So gilt eine Überexpression bei Epileptikern als Hauptursache für Therapieversagen (s. u.: Klinische Bedeutung von P-gp).
Pharmakogenetik von P-gp und MRP2
Polymorphismen im ABCB1-Gen waren Gegenstand einer Vielzahl von Untersuchungen. Derzeit besteht weitgehend Konsens, dass die Bioverfügbarkeit von Arzneistoffen im Plasma durch P-gp-Varianten nur wenig beeinflusst wird. Die Rolle der ABCB1-Varianten für die intrazellulären Konzentrationen, z. B. in Lymphozyten, oder für den Transport über die Blut-Hirn-Schranke wird diskutiert.
Bestimmte Polymorphismen im ABC-Transporter MRP2 (ABCC2) führen zu einer Erhöhung der Bioverfügbarkeit z. B. von Methotrexat. Aufgrund der hohen Variabilität der MRP2-Expression kann dieses Merkmal aber nicht für die Vorhersage der Aktivität genutzt werden. Ein erblicher Defekt von MRP2 führt zum Dubin-Johnson-Syndrom. Hepatozyten können hier nur vermindert Bilirubin-Derivate in die Galle ausscheiden, es kommt zur schwarzen Pigmentierung der Leber.
Während beim Menschen keine inaktive Variante von P-gp bekannt ist, weisen bestimmte Hunderassen wie Collies eine komplette Inaktivität von P-gp auf und reagieren deshalb mit einer erhöhten Empfindlichkeit auf bestimmte Arzneimittel (u. a. Antiparasitika wie Ivermectin, aber auch Opioide).
Klinische Bedeutung von ABCB1
Durch sein sehr großes Substratspektrum kann P-Glykoprotein an verschiedenen unerwünschten Arzneimittelwirkungen beteiligt sein. Es kann auch zwischen Arzneistoffen, die Substrate bzw. Induktoren oder Inhibitoren des ABCB1 sind, zu Interaktionen kommen. Ein Beispiel ist das Herzglykosid Digoxin, ein P-gp-Substrat, das nur in vernachlässigbarem Maße metabolisiert wird. Die Bioverfügbarkeit einer oralen Digoxindosis ist maßgeblich von der Aktivität des P-gp im Dünndarm abhängig, weil P-gp das Digoxin nach Aufnahme in die Enterozyten sofort wieder ins Darmlumen eliminiert. Demnach schwächen Inhibitoren von P-gp die Resorptionsbarriere und erhöhen die Serumkonzentration von Digoxin. Beispiele sind die Kombination von Digoxin mit dem Antiarrhythmikum Chinidin oder dem Calciumantagonisten Verapamil. Die erhöhte Plasmakonzentration des Digoxins kann das Risiko kardialer (ventrikuläre Extrasystolen, Kammerflimmern) oder neurotoxischer Nebenwirkungen (Schwindel, Farbsehen, Gesichtsausfälle) erhöhen.
Ritonavir hemmt u. a. CYP3A4 und P-gp. Deshalb wird Ritonavir mit zwei anderen HIV-Medikamenten, die CYP3A4- und P-gp-Substrate darstellen, fix kombiniert, um deren Wirkkonzentrationen über einen längeren Zeitraum in den Lymphozyten zu erhöhen; Ritonavir fungiert hier als „Booster“.
Vorsicht vor P-gp-Inhibitoren ist auch bei nicht-verschreibungspflichtigen Substanzen geboten. So wurde 2008 vor dem Missbrauch des Antidiarrhoikums Loperamid in der Drogenszene gewarnt, denn mit zwei einfachen „Tricks“ kann man dieses Opioid ins zentrale Nervensystem befördern: Durch eine inhalative oder sublinguale Applikation werden hohe Plasmaspiegel erreicht (Umgehung des First-pass-Effekts), und durch die vorherigen Einnahme von P-gp-Inhibitoren und CYP3A4-Inhibitoren wie Grapefruitsaft oder Chinin wird der Abbau verlangsamt. Die so ausgelösten ZNS-Effekte waren von der Stärke her vergleichbar mit Fentanyl. Entsprechende Anleitungen zum „Scharfmachen“ von Loperamid sind auf einschlägigen Seiten im Internet zu finden. Chinin wurde in Deutschland am 1. April 2015 der Verschreibungspflicht unterstellt. Diese Entscheidung wurde allerdings nicht aufgrund seines Missbrauchspotenzials getroffen, sondern aufgrund der Risiken für Thrombozytopenien und Herzrhythmusstörungen sowie des beachtlichen Interaktionspotenzials (s. DAZ 2014, Nr. 17, S. 32).
P-gp beeinflusst auch die Bioverfügbarkeit gerinnungshemmender Medikamente. Während bei Vitamin-K-Antagonisten eine gleichzeitige Anwendung mit starken P-gp-Inhibitoren prinzipiell möglich ist, da der INR-Wert engmaschig überwacht und die Dosis angepasst werden kann, ist die Kombination mit den direkten Gerinnungshemmern Dabigatran, Apixaban und Rivaroxaban, bei denen derzeit keine Labortests für die Gerinnungsfunktion existieren, kontraindiziert.
Umgekehrt kann es aber auch vorteilhaft sein, wenn ein Arzneistoff ein gutes P-gp-Substrat ist. Cetirizin, ein bekanntermaßen wenig sedierendes Antihistaminikum der zweiten Generation, ruft so wenige ZNS-Nebenwirkungen hervor, weil es als P-gp-Substrat kaum die Blut-Hirn-Schranke passiert.
Die Induktion von P-gp reduziert die Bioverfügbarkeit von Arzneistoffen durch deren beschleunigten Auswärtstransport. So vermindert Hyperforin, ein Inhaltsstoff des Johanniskrauts und potenter P-gp-Induktor, bei gleichzeitiger Einnahme hormoneller Kontrazeptiva deren empfängnisverhütende Wirkung. Außer einem verstärkten Auftreten von Zwischenblutungen sind auch einige Fälle von ungewollter Schwangerschaft dokumentiert. P-gp-Induktoren wie das Tuberkulostatikum Rifampicin sollen nicht gleichzeitig mit Ciclosporin eingenommen werden, denn sie verhindern, dass ausreichende therapeutische Konzentrationen von Ciclosporin erreicht werden. Weitere P-gp-induzierende Medikamente finden sich in Tabelle 2.
Solute Carriers (SLC)
Die meisten SLC dienen überwiegend der zellulären Aufnahme (Influx) von Nährstoffen oder der Verteilung von Gallensäuren oder Hormonen. Sie kommen im ganzen Körper vor und werden überwiegend durch den Natriumgradienten angetrieben. Die SLC-Familien werden mit einer Zahl bezeichnet (s. Tab. 1). Auch hier sind noch alte Nomenklaturen in Gebrauch wie organic anion/cation transporter (OAT/OCT = SLC22) oder organic anion-transporting polypeptide (OATP = SLCO = SLC21) sowie Funktionsnamen wie peptide transporter (PEPT = SLC15) oder urate transporter (URAT = SLC22). Die einzelnen SLC tragen zusätzlich zum Familiennamen einen Buchstaben und eine Zahl.
Pharmakokinetisch relevante SLC befinden sich vor allem in Leber und Niere. SLC sind aber ubiquitär in Zellmembranen, Vesikelmembranen und der mitochondrialen Doppelmembran vorhanden. Ein bekanntes physiologisches Beispiel ist SLC5A1, ein Natrium-Glucose-Cotransporter. Zur Flüssigkeitssubstitution bei Patienten mit Durchfallerkrankung müssen Salz und Zucker gemeinsam gegeben werden: Glucose und Natrium werden so in einem gemeinsamen Transportvorgang im stöchiometrischen Verhältnis 1:1 aufgenommen. Dadurch verschiebt sich der osmotische Gradient, und Wasser kann wieder aus dem Darm in den Blutkreislauf diffundieren. Cola-Getränke und Salzstangen sind nicht für die Rehydratisierung geeignet, da aufgrund des ungünstigen Zucker-Salz-Verhältnisses (zu viel Zucker, zu wenig Salz) sogar Wasser aus dem Körper in den Darm gezogen werden könnte. Daher sollte immer die WHO-Trinklösung, z. B. Oralpädon® oder Elotrans®, angewendet werden.
SLC können auch Zielstrukturen für Medikamente sein: Diuretika hemmen renale Transporter, Antidepressiva hemmen neuronale Monoamintransporter (s. Kasten „SLC als pharmakologische Zielstrukturen“).
SLC als pharmakologische Zielstrukturen
Wie die Enzyme des Arzneistoffwechsels können auch Transporter als pharmakodynamische Zielstrukturen genutzt werden:
Dapagliflozin ist ein neues Antidiabetikum aus der Gruppe der SGLT2-Inhibitoren. SGLT2, auch bekannt als SLC5A2, ist ein Natrium/Glucose-Cotransporter, der – im Unterschied zu SLC5A1 – vor allem renal exprimiert wird und die Wiederaufnahme von Glucose aus dem Urin vermittelt. Durch Hemmung von SGLT2 kommt es zu einer verstärkten Glucoseausscheidung und nachfolgend zu einer Senkung des Blutzuckerspiegels.
Die SLC-Familien 1, 6, 17, 18 und 32 regulieren die Wiederaufnahme von Neurotransmittern aus dem synaptischen Spalt. Dementsprechend sind z. B. zahlreiche Antidepressiva sowie Arzneistoffe zur Behandlung des Aufmerksamkeits-Defizit-Hyperaktivitäts-Syndroms Hemmstoffe des Norepinephrin-Transporters (NET = SLC6A2). Die selektiven Serotonin-Wiederaufnahmehemmer (SSRI) hemmen vor allem SERT = SLC6A4.
Die Zielstrukturen einiger Diuretika sind ebenfalls SLC: Furosemid hemmt einen Na/K/2Cl-Cotransporter (NKCC2 = SLC12A1), Hydrochlorothiazid hemmt einen Na/Cl-Cotransporter (NCC = SLC12A3).
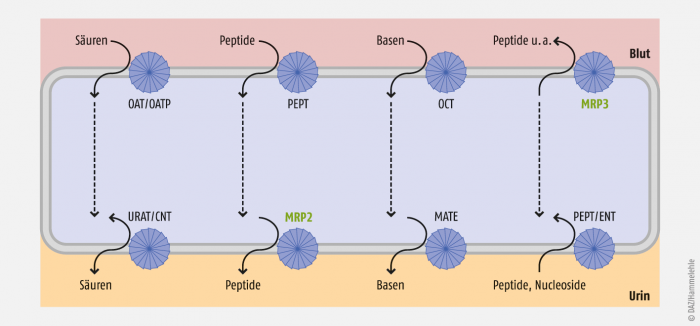
Probenecid und Benzbromaron sind Inhibitoren der Transporter OAT (basolateral) und hURAT1 (luminal), die in der Niere sowohl die Ausscheidung als auch die Rückresorption von Harnsäure vermitteln (Abb. 3). Abhängig von der Konzentration des Inhibitors wirkt dieser somit urikosurisch oder „paradox“ harnsäureretinierend. Die Nephrotoxizität von Cidofovir kann durch die gleichzeitige Gabe von Probenecid stark reduziert werden.
Abb. 3: Nierenzelle mit einigen typischen Transportern an der basolateralen (oben) und luminalen Zellmembran (unten). Dargestellt sind ABC-Transporter (MRP) und SLC (alle anderen), die synergistisch miteinander operieren. Sie können sowohl Substrate in den Urin ausscheiden als auch durch einen „körperwärts“ gerichteten Transport aus dem Urin rückresorbieren.
Wie die Beispiele URAT und CNT (concentrative nucleoside transporter, SLC28) zeigen, können SLC auch Effluxtransporter sein (Abb. 3). Die Transportrichtung eines SLC hängt vor allem von dem jeweiligen Organ ab, aber der ENT (equilibrative nucleoside transporter, SLC29) kann grundsätzlich in beide Richtungen (einwärts und auswärts) transportieren.
Pharmakogenetik von SLC
Bei der seltenen Hyperbilirubinämie vom Rotor-Typ (auch Rotor-Syndrom) sind weder OATP1B1 (syn. SLCO1B1, SLC21A6, OATP-C) noch OATP1B3 funktionsfähig. Die Störung führt zur Gelbsucht, ist aber relativ harmlos.
Eine genetisch bedingte Einschränkung der Funktion von OAPT1B1 kann zu einer erhöhten Empfindlichkeit für unerwünschte Wirkungen (UAW) einer Statintherapie beitragen. So weisen homozygote Träger der OATP1B1-Variante 431C→T ein 17-fach erhöhtes Risiko für eine Myopathie (Muskelschmerzen bis hin zur Rhabdomyolyse) unter 80 mg Simvastatin auf. Die Europäische Arzneimittelagentur EMA hat kürzlich diese Information in die Produktbeschreibung von Simvastatin aufgenommen.
Klinische Bedeutung von SLC
SLC können durch bestimmte Arzneistoffe inhibiert werden, dagegen sind klinisch relevante Induktoren nicht bekannt (Tab. 3).
| Transporter | Substrate | Inhibitoren |
|---|---|---|
| OATP1B (SLC21-Familie) | ACE-Inhibitoren/AT1 -Antagonisten wie Telmisartan (OATP1B3)BosentanCiclosporinEstradiolEzetimibFexofenadinGlinide, z. B. Nateglinid und RepaglinidGlitazone, z. B. Pioglitazon LopinavirMethotrexatMycophenolatRifampicinSirolimusSN-38 (Irinotecan-Metabolit)Statine wie SimvastatinTorasemidGifte: Arsen, Phallotoxine/Amatoxine, Microcystin-LR | CiclosporinCimetidinGemfibrozilGlibenclamidMakrolide wie ClarithromycinMetformin PenicillinProteaseinhibitoren, z. B. Ritonavir, LopinavirRifampicinTacrolimusInhaltsstoffe in Zitrusfrüchten, Äpfeln und grünem TeeSilibilin (Mariendistel) |
| OATP2B1 (SLC21A9) | ähnlich OATP1B, aber vor allem: Benzylpenicillineinige Statine, z. B. Atorvastatin | ähnlich OATP1B, aber vor allem: Ciclosporin |
| OCT (SLC22-Familie) | MetforminNucleosidanaloga | AmprenavirDipyrimadolKetoconazolProbenecid |
| OAT und hURAT1 (SLC22-Familie) | NSAIDβ-Lactam-Antibiotika, Ciprofloxacin | BenzbromaronProbenecid |
| MATE1 (SLC47) | AciclovirCephalexinCimetidinMetforminSirolimus | Azol-AntimykotikaCimetidinCiprofloxacinDipyridamolImatinibRapamycinRitonavirTrimethoprim |
Beim „Lipobay®-Skandal“ (Wirkstoff Cerivastatin) im Jahr 2001 wurde weltweit die große Bedeutung des durch OATP1B1 vermittelten Transports von Pharmaka in die Leberzellen erkannt. Zuvor war bekannt gewesen, dass Statine bei Überdosierungen eine Rhabdomyolyse auslösen können und dass diese UAW bei gleichzeitiger Therapie mit Gemfibrozil gehäuft auftrat. Der molekulare Mechanismus konnte jedoch erst in den Folgejahren aufgeklärt werden: Gemfibrozil ist ein OATP-Inhibitor (s. Tab. 3); bei Kombination mit einem Statin verhindert es, dass das Statin in die Leberzelle gelangt. Stattdessen steigt die Plasmakonzentration des Statins, und es kann den Muskelmetabolismus stören. Erschwerend kommt noch hinzu, dass Gemfibrozil CYP2C8, welches Cerivastatin abbaut, hemmt.
Auch Ciclosporin hemmt OATP1B1 und darf nicht zusammen mit Statinen eingenommen werden. Dabei hängt der Anstieg der AUC der einzelnen Statine von ihrer hepatischen Clearance ab: Die Bioverfügbarkeit von Fluvastatin ist z. B. nur wenig von Arzneimittelinteraktionen oder pharmakogenetischen Varianten betroffen.
Methotrexat (MTX) ist ebenfalls ein SLC-Substrat, es wird z. B. vom Folattransporter (SLC19A1) in die Zellen aufgenommen, und weitere SLC sind an seiner renalen Elimination beteiligt. Weil Acetylsalicylsäure oder andere NSAID diese SLC hemmen, können sie bei einer MTX-Therapie dessen Plasmakonzentration erhöhen, weshalb diese Kombination laut Fachinformation kontraindiziert ist. Grippeähnliche Symptome und dunkler Urin können hier ein Hinweis auf diese Interaktion sein. Die Elimination von MTX wird auch durch Penicilline (v. a. Piperacillin) stark eingeschränkt.
Aufgrund der großen therapeutischen Breite von Penicillinen und Cephalosporinen, die Substrate für OAT/OATP sind, sind Interaktionen nicht klinisch relevant. Die antibiotische Wirkung kann im Einzelfall mit OAT-Inhibitoren wie Probenecid gesteigert werden, doch stellt dieser Booster im Unterschied zum Ritonavir bei HIV-Medikamenten (s. o.) kein Standardvorgehen dar!
Lithium wird renal eliminiert und konkurriert mit Natrium um die tubuläre Wiederaufnahme, d. h. um die entsprechenden Transporter. Schleifendiuretika wie Furosemid, die über kompensatorische Mechanismen die tubuläre Wiederaufnahme von Natrium steigern, bewirken auch eine stärkere Rückresorption von Lithium, was zu erhöhten Lithiumplasmakonzentrationen führt.
Die SLC-Inhibitoren Penicillin und Silibinin (aus der Mariendistel) können bei Knollenblätterpilzvergiftung als Antidote verwendet werden, da sie die Aufnahme von Amatoxinen in die Leberzellen blockieren.
Transporter-bedingte Interaktionen bei der antiretroviralen Therapie
Insbesondere bei der Behandlung von Infektionen mit Humanem Immunodefizienzvirus (HIV) und Hepatitis-C-Virus (HCV) treten regelmäßig starke pharmakokinetische Arzneimittelinteraktionen auf. Die meisten Substanzen sind gleichzeitig Substrat, Inhibitor und Induktor von Enzymen der Phase I, Phase II und Phase III: Proteaseinhibitoren (PI) sind Inhibitoren von CYP-Isoenzymen und P-gp; nichtnucleosidische Reverse-Transkriptase-Inhibitoren (NNRTI) sind Inhibitoren von ABC-Transportern und SLC (s. Tab. 4).
| Substanz | P-gp | MRP- | BCRP | OATP- | OCT1/2 | OAT1/3 | CNT1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 1A2 | 1B1 | 1B3 | 2B1 | ||||||
Proteasehemmer | |||||||||||
| Ritonavir | S H > I | S H | S H > I | H | H | H > I | H > I | H | H | ||
| Lopinavir | S H > I | S | S | H | S | S H | H | H | |||
| Saquinavir | S H > I | S H | S | H | S H | S H | H | H | |||
| Indinavir | S | S H | S | H | H | H | |||||
Nucleosidische Reverse-Transkriptase-Hemmer (NRTIs) und Nicht-nucleosidische Reverse-Transkriptase-Hemmer (NNRTIs) | |||||||||||
| Lamivudin, NRTI | H > I | H | H | S | S H | H | S H | ||||
| Emtricitabin, NRTI | H > I | S H | S H | ||||||||
| Tenofovir, NRTI | S H | H | H | S | S | ||||||
| Efavirenz, NNRTI | I H | H > I | H | H | |||||||
| Nevirapin, NNRTI | I H | H | H | H | |||||||
Sonstige | |||||||||||
| Raltegravir | S | ||||||||||
| Maraviroc | S H | ||||||||||
Diese komplexen Interaktionen werden in Leitlinien und Fachinformationen berücksichtigt. So machen z. B. die Fachinformationen des CCR5-Antagonisten Maraviroc sehr detaillierte Angaben über die inhibitorischen und induktiven Eigenschaften der Substanz, in vivo und in vitro; zudem gibt eine Tabelle Hinweise, wenn Maraviroc zusätzlich zu einer bestehenden antiretroviralen Therapie gegeben werden soll.
Da etwa 60 Prozent aller HIV-Patienten auch HCV-positiv sind, sind Kombinationen aus diesen beiden Arzneistoffgruppen ebenfalls erforscht und gut dokumentiert. Dennoch werden manchmal unerwartete Interaktionen bekannt:
Fallbeispiel
Am 23. 2. 2012 wurde ein Rote Hand-Brief zu dem HCV-Medikament Boceprevir veröffentlicht. Unerwartet wurden unter Therapien mit Lopinavir/Ritonavir und Boceprevir Abnahmen der Plasmakonzentrationen aller drei Medikamente von ca. 40 bis 50 Prozent beobachtet. Ritonavir ist ein starker Inhibitor von CYP-Enzymen und P-gp. Über Boceprevir war lange Zeit v. a. die starke inhibitorische Wirkung auf CYP3A4 bekannt, ebenso dass es ein Substrat von P-gp und BCRP sei. Demnach hätte bei Kombination dieser hemmenden Arzneistoffe das Gegenteil, nämlich eine Erhöhung der Plasmakonzentrationen aller drei Medikamente, erfolgen sollen.
Hulskotte et al. wiesen darauf hin, dass Ritonavir ein starker Inhibitor von OATP und OCT ist, was zumindest den Plasmakonzentrationsabfall von Boceprevir erklären kann, denn es wird über diverse SLC-Transporter aus dem Gastrointestinaltrakt resorbiert [2].
Chu et al. zeigten in vitro, dass Boceprevir u. a. OAPT1B-Isoenzyme hemmt [8]. Dieser Mechanismus könnte die verminderte Plasmakonzentration von Lopinavir erklären, weil dieses u. a. über OATP1B1 resorbiert wird (Tab. 3). |
Literatur
[1] www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2012/20120223.pdf
[2] Hulskotte EG, et al. Pharmacokinetic interactions between the hepatitis C virus protease inhibitor boceprevir and ritonavir-boosted HIV-1 protease inhibitors atazanavir, darunavir, and lopinavir. Clin Infect Dis 2013;56(5):718-26
[3] Cascorbi I. Arzneimittelinteraktionen: Prinzipien, Beispiele und klinische Folgen. Dtsch Arztebl Int 2012;109(33-34):546-56
[4] Cascorbi I. P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb Exp Pharmacol 2011;201:261-83
[5] BioParadigms. SLC Tables; http://slc.bioparadigms.org
[6] Kis O, et al. The complexities of antiretroviral drug-drug interactions: role of ABC and SLC transporters. Trends Pharmacol Sci 2010;31(1):22-35
[7] Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005;2(1):86-98
[8] Chu X, et al. In vitro assessment of drug-drug interaction potential of boceprevir associated with drug metabolizing enzymes and transporters. Drug Metab Dispos 2013;41(3):668-681
[9] Herdegen T. Kurzlehrbuch Pharmakologie und Toxikologie, 3. Aufl. Stuttgart 2013
Autoren
Dr. rer. nat. Kirstin Reinecke, Dr. med. Ruwen Böhm, Prof. Dr. med. Thomas Herdegen, Prof. Dr. med. Dr. rer. nat. Ingolf Cascorbi
Institut für Experimentelle und Klinische Pharmakologie
Arnold-Heller-Str. 3, Haus 30, 24105 Kiel
Foto: lofik – Fotolia.com
Rückblick
Mit diesem Beitrag endet diese DAZ-Serie über Arzneimittelinteraktionen, in der zuvor die folgende Beiträge erschienen waren:
- Arzneimittelinteraktionen verstehen, vermitteln und vermeiden. DAZ 2012, Nr. 36, S. 64 – 74
- Interaktionen mit CYP3A4. DAZ 2012, Nr. 40, S. 58 – 67
- Arzneimittel und CYP2D6. DAZ 2012, Nr. 47, S. 60 – 66
- Arzneimittel und CYP1A2. DAZ 2013, Nr. 22, S. 44 – 49
- CYP2C und CYP2E1. DAZ 2014, Nr. 30, S. 48 – 52



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.