










- DAZ.online
- DAZ / AZ
- DAZ 47/2012
- Arzneimittel und CYP2D6
Klinische Pharmazie
Arzneimittel und CYP2D6
Vor wenigen Wochen haben FDA und BfArM gewarnt, dass einige Kleinkinder nach Gabe von Codein hohe Plasmakonzentrationen des aktiven Metaboliten Morphin erreicht haben, die teilweise zu tödlichen Atemdepressionen geführt haben. Derzeit geht man in den beschriebenen Fällen von einer erhöhten Umsetzungsgeschwindigkeit von Codein zu Morphin aus. Diese Reaktion wird durch CYP2D6 katalysiert.
CYP2D6 metabolisiert – zumindest anteilsmäßig – über 20% aller durch Phase-1-Reaktionen metabolisierten Arzneistoffe. Dies entspricht in etwa 15% aller auf dem Markt befindlichen Arzneistoffe (s. DAZ 2012; Nr. 36, S. 64). Aufgrund seiner eher geringen hepatischen Expression (ca. 2%) wird in vivo eine etwas geringere Relevanz vermutet, als In-vitro-Experimente suggeriert haben.
So hat Amitriptylin beispielsweise in vitro eine etwa gleich gute Affinität zu CYP2D6 und CYP2C19. Da aber CYP2C19 deutlich stärker hepatisch exprimiert wird (bis zu ca. 18%) als CYP2D6, geht man davon aus, dass CYP2C19 beim Abbau von Amitriptylin in vivo eine größere Bedeutung hat als CYP2D6. Tatsächlich konnte die Amitriptylinkonzentration nur in 20% der Fälle, die metabolic ratio Amitriptylin / 10-OH-Amitriptylin nur in 45% allein durch die CYP2D6-Aktivität erklärt werden [2]. Die – rein rechnerische – Dosisanpassung in Abhängigkeit von der CYP2D6-Aktivität liegt bei Amitriptylin zwischen 75 und 130% der Standarddosis [8].
Arzneistoffe, die fast ausschließlich von CYP2D6 abgebaut werden, benötigen hingegen stärkere Dosisanpassungen (z. B. 25% bis 180% der Standarddosis von Imipramin, je nach dem CYP2D6-Phänotyp). Ob auch der therapeutische Erfolg direkt mit diesen pharmakokinetischen Überlegungen korreliert, konnte bis heute jedoch nicht gut gezeigt werden, zumal der aktive Metabolit Nortriptylin als CYP2D6-Substrat auch Einfluss auf die Wirkung hat.
CYP2D6 wird auch im Darm, Gehirn, in der Lunge und anderen extrahepatischen Geweben exprimiert. Es verstoffwechselt keine essenziellen endogenen Substanzen – eine notwendige Voraussetzung dafür, dass es individuell sehr unterschiedlich exprimiert wird, d. h. dass im menschlichen Genpool diverse Genvarianten vorhanden sind, die zu unterschiedlichsten Enzymaktivitäten führen (Tab. 1): Einige Menschen verfügen z. B. über gar kein funktionsfähiges CYP2D6. Am Beispiel des CYP2D6-Polymorphismus lassen sich die Variationsbreite der individuellen Verträglichkeit und Wirksamkeit von Pharmaka anschaulich darstellen.
Von anderen CYP-Isoenzymen unterscheidet sich CYP2D6 nicht nur durch seine ausgeprägte pharmakogenetische Variabilität, sondern auch dadurch, dass es kaum induzierbar ist. Somit spielen nur Inhibitoren und Substrate von CYP2D6 eine Rolle für Arzneimittelinteraktionen.
Pharmakogenetik von CYP2D6
Seit der Entdeckung interindividueller Unterschiede in der Metabolisierungskapazität von CYP2D6 in den 70er Jahren zählt es zu den am besten charakterisierten Isoenzymen. Die sehr häufig auftretenden Polymorphismen führen zu stärker beziehungsweise schwächer aktiven oder sogar funktionslosen Varianten des Enzyms. Beim Menschen lässt sich durch Verabreichung spezieller Modellsubstanzen wie z. B. dem Antitussivum Dextromethorphan die Metabolisierungsaktivität von CYP2D6 charakterisieren (Phänotypisierung).
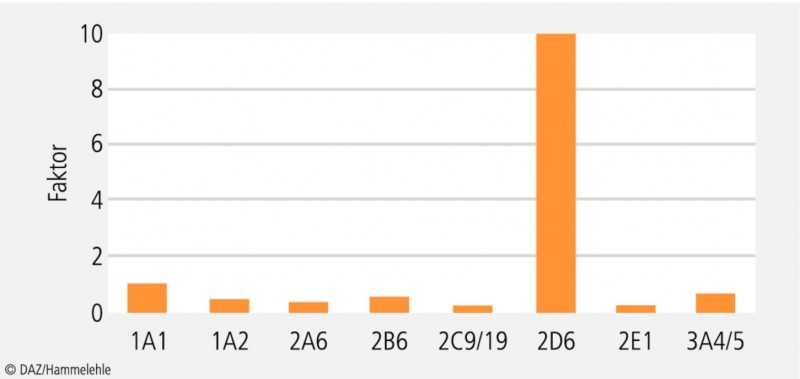
Die Eliminationsgeschwindigkeit von CYP2D6-Substraten kann sich bis zum Faktor 100 unterscheiden (Abb. 2). Grundsätzlich werden hinsichtlich der Metabolisierungsaktivität von CYP2D6 vier Typen unterschieden (Tab. 1):
Langsame Metabolisierer (poor metabolizer, PM) besitzen zwei nicht funktionelle Allele. Damit wird kein CYP2D6 gebildet und der Metabolismus der Substrate verläuft extrem langsam. Schon bei Standarddosierung ist mit der Akkumulation von Substrat-Wirkstoffen zu rechnen. Überdurchschnittlich hohe Plasmaspiegel sind die Folge mit verstärkten Nebenwirkungen, Intoxikationen und Interaktionen. Bei einem Prodrug, das durch CYP2D6 aktiviert wird, werden andererseits keine ausreichenden Wirkstoffspiegel erzielt.
Intermediäre Metabolisierer (intermediate metabolizer, IM) besitzen entweder zwei Genkopien, die für ein Enzym mit reduzierter Aktivität kodieren (homozygot) oder ein normales und ein dysfunktionales Allel (heterozygot). Pharmaka werden demnach nur mit reduzierter Aktivität verstoffwechselt. Die resultierenden pharmakologischen Effekte ähneln denen der PM in abgeschwächter Form.
Extensive (normale) Metabolisierer (extensive metabolizer, EM) sind homozygot für das Wildtyp-Allel (normale Allelform), d. h. es werden voll funktionsfähige CYP2D6-Proteine in physiologischer Konzentration gebildet. Arzneistoffe werden bei diesem Phänotyp effizient metabolisiert, die Wirkung des Arzneistoffes entspricht den Erwartungen an eine Standarddosierung.
Ultraschnelle Metabolisierer (ultrarapid metabolizer, UM) weisen aufgrund einer Genamplifikation drei oder mehr Kopien funktionsfähiger Allele auf. Betroffene Patienten metabolisieren Arzneistoffe so schnell, dass wirksame Plasmaspiegel nicht oder nur für kurze Zeit erreicht werden können. Sie sprechen selbst nach Gabe sehr hoher Dosen auf die Pharmakotherapie nicht an. Demgegenüber wird bei der Applikation eines Prodrugs pro Zeiteinheit so viel der aktiven Wirkform gebildet, dass toxische Konzentrationen erreicht werden können bzw. unerwünschte Arzneimittelwirkungen auftreten.
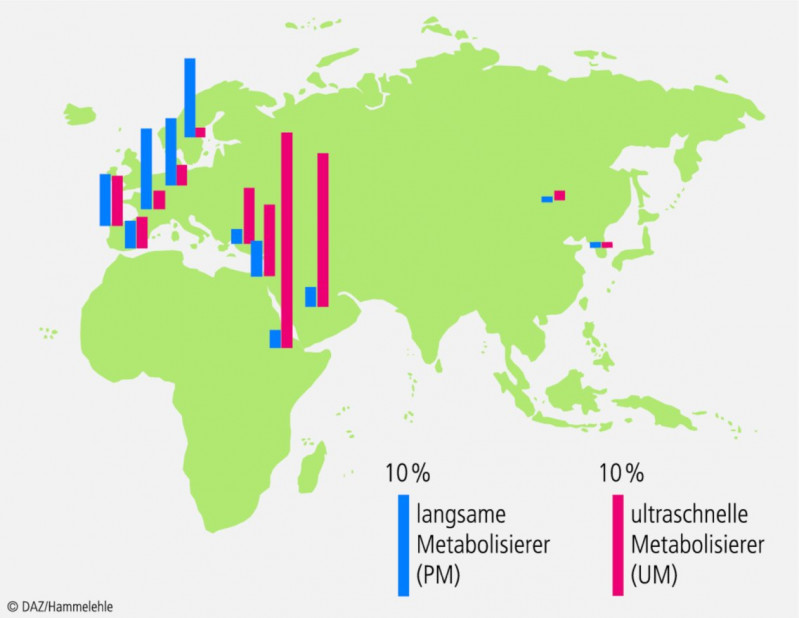
Die verschiedenen CYP2D6-Genotypen und -Phänotypen zeigen eine ausgeprägte geografische Häufung (Abb. 1, Tab. 1). Im Norden Europas gibt es wesentlich mehr langsame Metabolisierer als in Afrika oder dem nahen Osten, wo Genduplikationen und dementsprechend ultraschnelle Metabolisierer überwiegen.
Inhibition bei Varianten
Aufgrund der starken pharmakogenetischen Variabilität kann die Stärke einer Arzneimittelinteraktion unterschiedlich relevant sein: Während bei langsamen Metabolisierern die Medikation mit einem Inhibitor von CYP2D6 keinerlei Veränderungen nach sich zieht (kein Enzym vorhanden → keine Inhibition möglich), sind bei einem ultraschnellen Metabolisierer extrem große Veränderungen der CYP2D6-Aktivität zu erwarten (viel Enzym vorhanden → alles wird inhibiert).
Substrate von CYP2D6
Klinisch relevante Substrate von CYP2D6 sind
zahlreiche kardiovaskuläre Arzneimittel (Betablocker, Antiarrhythmika),
Neuro- und Psychopharmaka (v. a. Dibenzazepine wie die trizyklischen Antidepressiva, klassische Neuroleptika und Monoaminoxidasehemmer),
die Opioide Tramadol, Codein und Dextromethorphan sowie
der Estrogeninhibitor Tamoxifen (Tab. 2).
Zur Beurteilung der klinischen Relevanz müssen sowohl das Ausmaß der Metabolisierung über CYP2D6 bei den einzelnen Wirksubstanzen als auch die klinische Wertigkeit der Unterschiede in der polymorphen Metabolisierung berücksichtigt werden. Zusätzlich spielen Faktoren wie beispielsweise die äquipotente Aktivität der Muttersubstanz und deren Metaboliten ebenso eine Rolle wie die therapeutische Breite des Arzneistoffes oder die Möglichkeit alternativer Abbauwege.
Wirkungsverlust von Tamoxifen durch CYP2D6-InhibitorenTamoxifen ist ein weit verbreiteter Aromatase-Inhibitor, der bei hormonsensiblen Mammakarzinomen die Inzidenz und das Wachstum von Brustkrebs signifikant vermindert. Tamoxifen wird mittels CYP2D6 in zwei Schritten in seine aktiven Wirkform Endoxifen überführt. Bei langsamen CYP2D6-Metabolisierern ist neben der Bildung von Endoxifen auch der Therapieerfolg von Tamoxifen vermindert [9]. Zusätzlich besitzt die Komedikation mit CYP2D6-Inhibitoren das klinisch relevante Risiko einer Wirkungsabschwächung. Daher ist die Komedikation mit SSRI-Antidepressiva, und insbesondere mit Paroxetin, kontraindiziert. Eine aktuelle Metaanalyse [7] hat neben dem CYP2D6-Phänotyp und der Komedikation von CYP2D6-Inhibitoren noch weitere Faktoren zusammengetragen, die die therapeutische Wirkung von Tamoxifen beeinflussen:
! Als nicht CYP2D6-inhibierende Antidepressiva können Escitalopram, Citalopram, Venlafaxin oder Mirtazapin eingesetzt werden. |
Muttersubstanz aktiv, Metaboliten inaktiv
Das OTC-Antitussivum Dextrometorphan (DMP) wird hauptsächlich über CYP2D6 in den aktiven Metaboliten Dextrorphan (DO) umgewandelt. Dieser besitzt eine höhere Affinität zum NMDA-Rezeptor und blockiert diesen stärker als die Muttersubstanz. Die Hemmung von NMDA-Rezeptoren birgt das Risiko für psychotische Nebenwirkungen ebenso wie für Missbrauch. Schnelle Metabolisierer (1 – 10% der Kaukasier) besitzen für diese Nebenwirkungen von DO ein erhöhtes Risiko. Andererseits können bei langsamen Metabolisierern (7 – 10% der Kaukasier) die Nebenwirkungen von DMP wie Schwindel, Dysphorie oder Müdigkeit verstärkt sein. Die antitussive Wirkung (Agonismus am sigma-Rezeptor) wird durch den CYP2D6-Metabolismus nicht beeinflusst.
Muttersubstanz nicht oder wenig aktiv, Metaboliten aktiv (Prodrugs)
Andererseits können langsame Metabolisierer Prodrugs wie z. B. Codein oder Tramadol nicht zum aktiven Wirkstoff umwandeln und somit keine schmerzlindernde Wirkung entfalten:
Codein ist über einen derzeit noch unbekannten Wirkmechanismus antitussiv wirksam. Durch CYP2D6 wird es zum antitussiv und analgetisch wirksamen Morphin aktiviert. Eine Hemmung von CYP2D6 verschiebt also das Wirkspektrum von "analgetisch und antitussiv" zu "nur antitussiv".
Beim Racemat Tramadol hemmt ein Enantiomer die Serotonin- und Noradrenalinwiederaufnahme. Erst seine aktiven Metaboliten, die u. a. CYP2D6-vermittelt entstehen, sind starke µ-Opioidrezeptoragonisten. Eine Hemmung von CYP2D6 könnte hier die analgetische Wirkung gefährden und die Gefahr von serotonergen UAW wie Übelkeit erhöhen.
Muttersubstanz und Metaboliten aktiv
Bei Antidepressiva aus der Gruppe der selektiven Serotonin-Reuptake-Inhibitoren (SSRI) und dem Opioid Dihydrocodein gibt es keine klinisch relevanten Unterschiede in Abhängigkeit von der CYP2D6-Aktivität, da diese Substanzen sowohl in unmetabolisierter Form wie auch als Metaboliten aktiv sind.
CYP2D6 im Gehirn
Zu den endogenen Substraten von CYP2D6 gehören das neuromodulatorische Hormon Progesteron (allosterischer GABAA-Modulator), endogene Morphinvorstufen, sowie diverse biogene Amine. CYP2D6 vermittelt z. B. die Umwandlung von Tyramin zu Dopamin und von 5-Methoxytryptamin zu Serotonin. CYP2D6 katalysiert hier jedoch nicht die primären Stoffwechselwege o. g. Substanzen, sodass die ausgeprägten pharmakogenetischen Unterschiede zu keinen starken Persönlichkeitsveränderungen führen.
Das aktive Zentrum von CYP2D6 bietet für kleine Moleküle mit tertiären oder aromatischen Aminogruppen sowie Benzolringen eine hohe Affinität [10]. Da ausgehend von diesen Molekülstrukturen sich zahlreiche endogene Neurotransmitter (z. B. aromatische Aminogruppe und Benzolring in Tryptamin-Derivaten), Neuro- und Psychopharmaka (tertiäre Aminogruppe in trizyklischen Antidepressiva, aromatische Aminogruppe in klassischen Antipsychotika) sowie psychotrope Substanzen, aber auch in der Peripherie kardiovaskulär wirksame Stoffe (sekundäre Aminogruppe und Benzolring in Betablockern) ableiten, ist es nicht überraschend, dass auch genau diese Substanzen das Gros der Substratliste ausmachen.
Effekte bei Opioidabhängigen
Eine Studie weist auf eine mögliche Assoziation von CYP2D6-Aktivität und Drogenkonsum hin: Bei Opioidabhängigen findet man verhältnismäßig wenig langsame CYP2D6-Metabolisierer. Umgekehrt sprechen abhängige langsame Metabolisierer besonders gut auf eine Opioidsubstitutionstherapie an, vermutlich da bei ihnen seltener die äußerst unangenehmen Entzugssymptome, verursacht durch einen schnellen Abfall der Gehirnkonzentration der psychotropen Substanz, auftreten [6].
Inhibition von CYP2D6
Häufig kann durch eine Monotherapie kein erwünschter Therapieerfolg erzielt werden, oder der Patient benötigt wegen einer neu aufgetretenen Erkrankung eine Komedikation. Bei jeder Form der Kombinationstherapie ist zu beachten, dass Arzneistoffe nicht nur Substrat für CYP2D6, sondern auch Inhibitor für dieses Isoenzym sein können (Tab. 3).
Besonderes Augenmerk muss auf die Verordnung von Neuropharmaka gerichtet werden. Insbesondere selektive Serotoninwiederaufnahme-Hemmer (SSRI) wie Fluoxetin und Paroxetin, die zugleich potente CYP2D6-Inhibitoren sind, bergen ein hohes Interaktionspotenzial. Die kombinierte Einnahme von CYP2D6-inhibierenden SSRI mit QT-Zeit-verlängernden trizyklischen Antidepressiva und Antipsychotika kann deren Wirkspiegel gefährlich erhöhen, bis hin zur Auslösung lebensbedrohlicher Herzrhythmusstörungen. Es sind sogar Todesfälle durch Fluoxetin-bedingte Intoxikationen mit Amitriptylin beschrieben. Die lange Halbwertszeit von Fluoxetin und Norfluoxetin (ca. 15 Tage) führt dazu, dass diese Interaktion manchmal übersehen wird.
Des Weiteren ist auch auf die kombinierte Gabe von SSRI mit bestimmten Betablockern wie z. B. Metoprolol (Tab. 3) zu achten, da auch hier der Abbau des Betablockers gehemmt wird mit unerwünschter Blutdrucksenkung und Bradykardie als mögliche Konsequenzen. Der Einsatz von SSRI bei der Tamoxifentherapie zur Depressionsbehandlung oder zur Unterdrückung des Flush als unerwünschte Arzneimittelwirkung ist ebenfalls problematisch.
Eine Nicht-Aktivierung von Prodrugs, die sich z. B. im Versagen einer Schmerztherapie äußern kann, muss im Zusammenhang mit der Gabe von CYP2D6-inhibierenden SSRI ebenfalls bedacht werden. Sichere Alternativen stehen mit Venlafaxin, Citalopram oder Escitalopram zur Verfügung. Hier sollte ein Austausch stattfinden.
Induktion von CYP2D6
Dem CYP2D6-Gen fehlen die Bindungsstellen für typische nukleäre Rezeptoren, die die Induktion anderer CYP-Isoenzyme vermitteln (z. B. PXR, CAR, AhR). Arzneimittelwechselwirkungen durch Induktoren spielen daher keine Rolle.
Der klinische Fall
Das Therapeutische Drug-Monitoring-Programm KONBEST (siehe DAZ 2012; Nr. 40, S. 66) bewertet ggf. auch den Einfluss von Arzneimittelinteraktionen auf gemessene unerwartete Plasmakonzentrationen. Im folgenden Fallbeispiel aus der KONBEST-Datenbank kann man die Auswirkung einer CYP2D6-Inhibition erkennen:
Bei einem 47-jährigen, 85 kg schweren Patienten (Nichtraucher, kein Kaffeetrinker) wurde die Plasmakonzentration von Risperidon bestimmt, da Bewegungsstörungen beobachtet worden waren. Der Patient bekam täglich 7 mg Risperidon sowie 1200 mg Valproat und 200 mg Levomepromazin (Tab. 4).
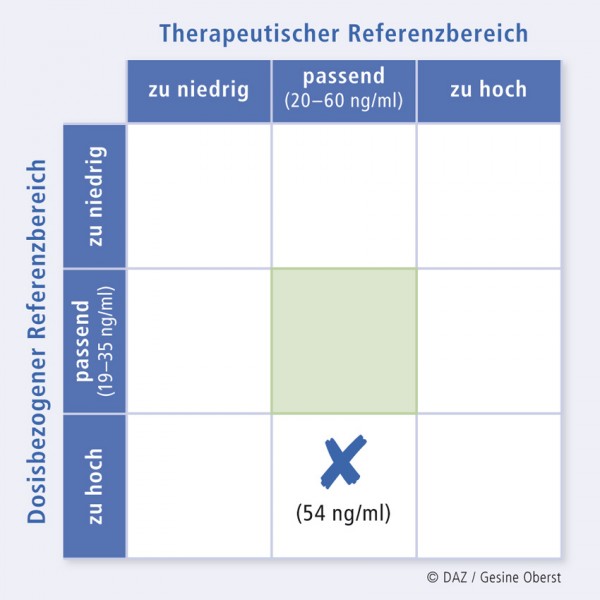
Die bestimmte Plasmakonzentration von Risperidon und seinem Metaboliten Paliperidon betrug 35 bzw. 19 ng/ml, also insgesamt 54 ng/ml; dieser Wert liegt zwar noch im therapeutischen Referenzbereich (20 – 60 ng/ml), aber weit oberhalb des bei dieser Dosis und diesem Körpergewicht erwarteten Bereiches von 19 bis 35 ng/ml liegt (Abb. 3).
Eine Erklärungsmöglichkeit ist die Hemmung der Risperidon-metabolisierenden Enzyme, nämlich CYP2D6 durch Levomepromazin und CYP3A4 durch Valproat (Tab. 4).
Das Wichtigste über CYP2D6 und Arzneimittel
|
Literatur
[1] Anzenbacher P, et al. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001;58(5-6):737-47.
[2] Baumann P, et al. Amitriptyline pharmacokinetics and clinical response: II. Metabolic polymorphism assessed by hydroxylation of debrisoquine and mephenytoin. Int Clin Psychopharmacol 1986;1(2):102-12.
[3] Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002;3(2):229-43.
[4] Cascorbi I. Pharmacogenetics of cytochrome P4502D6: genetic background and clinical implication. Eur J Clin Invest 2003;33 Suppl 2:17-22.
[5] Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine, 2007. http://medicine.iupui.edu/clinpharm/ddis/table.aspx.
[6] Haile CN, et al. Pharmacogenetic treatments for drug addiction: alcohol and opiates. Am J Drug Alcohol Abuse 2008;34(4):355-81.
[7] Hertz DL, et al. Tamoxifen and CYP2D6: a contradiction of data Oncologist 2012;17(5):620-30.
[8] Kirchheiner J, Seeringer A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim Biophys Acta 2007;1770(3):489-94.
[9] Schroth W, et al. Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA 2009;302(13): 1429-36.
[10] Zanger UM, et al. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol 2004;369(1):23-37.
Autoren
Dr. rer. nat. Kirstin Reinecke, Dr. med. Ruwen Böhm, Prof. Dr. med. Dr. rer. nat. Ingolf Cascorbi, Prof. Dr. med. Thomas Herdegen, Institut für Experimentelle und Klinische Pharmakologie, Arnold-Heller-Str. 3, Haus 30, 24105 Kiel, E-Mail: ruwen.boehm@pharmakologie.uni-kiel.de
Prof. Dr. med. Dr. rer. nat. Ekkehard Haen, Abteilung Klinische Pharmakologie/Psychopharmakologie Psychiatrische Universitätsklinik Regensburg Bezirksklinikum Regensburg, Universitätsstraße 84, 93053 Regensburg
RückblickBisher sind in dieser DAZ-Serie folgende Beiträge erschienen: 1. Arzneimittelaktionen verstehen, vermitteln und vermeiden. 2. Interaktionen mit CYP3A4. |



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.