
















- DAZ.online
- DAZ / AZ
- DAZ 37/2014
- Pharmakovigilanz in ...
Arzneimittelsicherheit
Pharmakovigilanz in Europa Der Ablauf eines Risikobewertungs-Verfahrens
Ein Großteil der sicherheitsrelevanten Informationen zu einem Arzneimittel muss zum Zeitpunkt der Marktzulassung vorliegen. Erst wenn basierend auf klinischen Daten zu Sicherheit und Wirksamkeit eine positive Nutzen-Risiko-Abschätzung wissenschaftlich gerechtfertigt ist, kann der Marktzugang erfolgen. Allerdings unterliegt die Risikobewertung zum Zeitpunkt der Zulassung einigen grundlegenden Limitationen: Die Zahl der Patienten in klinischen Studien ist begrenzt, so dass seltene und sehr seltene Nebenwirkungen oder bestimmte Patientengruppen mit erhöhtem Risiko für bestimmte Nebenwirkungen meist nicht vor der Zulassung erkannt werden können. Ein typisches Beispiel hierfür ist das kürzlich abgeschlossene europäische Risikobewertungsverfahren zum Risiko von Thrombosen nach der Einnahme oraler Kontrazeptiva. Hier kommen je nach Kontrazeptivum 5 bis 12 Fälle auf 10.000 Frauen, die die Pille ein Jahr lang eingenommen haben. Ohne Einnahme von Kontrazeptiva entwickeln aber auch bereits zwei von 10.000 Frauen innerhalb dieser Zeitspanne eine Thrombose. Übergewichtige und ältere Frauen tragen ein besonderes Risiko. Nach der Zulassung eines Arzneimittels müssen daher alle dem Zulassungsinhaber gemeldeten Vorkommnisse fortlaufend und systematisch im Hinblick auf die Arzneimittelsicherheit gesammelt und analysiert werden. Das oberste Ziel der Anwendung eines jeden Arzneimittels muss immer sein, dass der Nutzen seiner Anwendung die Risiken so weit wie möglich überwiegt.
Start eines Risikobewertungsverfahrens: Die Erkennung neuer Risiken
Im Rahmen der Arzneimittelüberwachung nach Markteinführung (Pharmakovigilanz) müssen nach Arzneimittelgesetz (AMG) Risiken für Patienten früh erkannt und weiterverfolgt werden. Dazu betreiben pharmazeutische Unternehmer und Arzneimittelbehörden unabhängig voneinander ein System zur frühzeitigen Erkennung neuer Risiken – die sogenannte Signaldetektion. Auslöser eines Risikobewertungsverfahrens können verschiedene Vorkommnisse sein:
- Häufung von Einzelfallberichten,
- Veröffentlichung neuer Studienergebnisse, die neue oder größere Risiken zeigen als bisher bekannt,
- Publikation anderer wissenschaftlicher Daten, die ein erhöhtes Risiko einer Nebenwirkung eines Arzneimittels zeigen,
- Mitteilung eines Zulassungsinhabers, dass ihm ein neues Risiko, welches im Zusammenhang mit seinem Arzneimittel stehen könnte, aufgefallen ist,
- Veröffentlichung von Warnhinweisen zu Arzneimitteln in Nicht-EU-Ländern,
- Marktrücknahme, Widerruf oder Nicht-Erneuern der Zulassung in einem EU-Staat.
Risiken müssen weiterverfolgt und bewertet werden, um Apotheker, Ärzte und Patienten auf dem aktuellsten Stand zu halten und gegebenenfalls Maßnahmen zu ergreifen diese Risiken zu minimieren (z.B. durch Anpassungen in den informativen Texten eines Arzneimittels). Von zentraler Bedeutung ist hierbei, in der Vielzahl der Informationen die relevanten von den nicht-relevanten Informationen zu trennen und wichtige Risiken zu erkennen. Diesen ersten Schritt der Risikobewertung übernehmen einerseits die Zulassungsinhaber selbst und andererseits unabhängige staatliche Institutionen. Bei Arzneimitteln, die nur in Deutschland, d.h. in keinem anderen EU-Staat zugelassen sind, sind alleine die regulatorischen Bundesoberbehörden zuständig und leiten gegebenenfalls ein Stufenplanverfahren ein. In Deutschland ist bei Impfstoffen, Blutprodukten oder Antikörpern das Paul-Ehrlich-Institut (PEI) zuständig und bei allen anderen Arzneimitteln das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM). Die Zuständigkeit für die Risikobewertung der meisten Arzneimittel, welche in allen oder mehreren EU-Mitgliedstaaten zugelassen sind, ist in der EU unter den Mitgliedsländern aufgeteilt. Das zuständige Land, der sogenannte „Rapporteur“, übernimmt EU-weit federführend die erste Bewertung sicherheitsrelevanter Aspekte (z.B. potenzielle Nebenwirkungen oder Arzneimittelinteraktionen etc.). Sämtliche neuen Informationen zu Risiken eines Arzneimittels werden über die EMA an den Rapporteur weitergeleitet, welcher zunächst eine Art Vorbewertung (Validierung) vornimmt. Hierbei wird geprüft, ob die neuen Informationen ausreichend sind um ein Verfahren zur Risikobewertung einzuleiten. Unabhängig davon sammelt und bewertet jedes EU-Land ebenfalls neue Erkenntnisse zu Arzneimitteln, und kann in gravierenden Fällen auch ohne Vorbewertung durch den Rapporteur direkt ein europäisches Risikoverfahren einleiten.
Referral-Verfahren: Bewertung grundsätzlicher Fragen zu Risiken
Die bekanntesten und wichtigsten Risikobewertungsverfahren sind die sogenannten Referral-Verfahren (von engl. referred: übertragen), welche der EMA die Aufgabe übertragen, die wissenschaftliche Bewertung eines Arzneimittels oder einer Gruppe von Arzneimitteln vorzunehmen. Im Folgenden soll daher der Begriff Referral mit dem Begriff europäisches Risikobewertungsverfahren gleichgesetzt werden.
Referral werden gestartet, wenn es in der EU um grundsätzliche Fragen zu Risiken von auf dem Markt befindlichen Arzneimitteln geht oder wenn gegensätzliche Entscheidungen einzelner Mitgliedsländer zu Arzneimitteln bestehen.
Es gibt unterschiedliche Arten von Referrals die auf der Leitlinie 2001/83/EC basieren (Fußnote zu finden unter: http://ec.europa.eu/health/documents/eudralex/vol-1/index_en.htm). Die wichtigsten sind:
- Artikel 29(4) bei divergierenden Meinungen von EU-Mitgliedstaaten hinsichtlich Gefahren für die öffentliche Gesundheit bei Verfahren der gegenseitig anerkannten oder initial dezentralen Zulassungen (DCP- und MRP-Verfahren)
- Artikel 30 bei der Notwendigkeit einer Harmonisierung der informativen Texte innerhalb der EU, also wenn beispielsweise unterschiedliche Indikationen innerhalb der Mitgliedstaaten gelten (umgangssprachlich: „Harmonisierungs-Referral“)
- Artikel 31 kommt zum Tragen wenn ein Gemeinschaftsinteresse an der Bewertung eines Risikos besteht, z.B. bei einem gut begründeten Verdacht auf ein schwerwiegendes neues Risiko („Community Interest referral“)
- Artikel-107i-Referrals sind beschleunigte Risikoverfahren bei besonderer Dringlichkeit z.B. wenn „Gefahr im Verzug“ ist (urgent Union procedure) (umgangssprachlich: „Pharmakovigilanz-Referral“)
Zudem sind zwei Arten von Referrals zu unterscheiden:
Referrals ohne Sicherheitsaspekte, welche direkt im Ausschuss für Humanarzneimittel (Committee for Medicinal Products for Human Use, CHMP) bewertet werden, sowie die große Gruppe der sicherheitsbezogenen Referrals, welche in die Zuständigkeit des Ausschusses für Risikobewertung im Bereich der Pharmakovigilanz der Europäischen Arzneimittelagentur, im englischen Pharmacovigilance Risk Assessment Committee (PRAC), fallen.
Der PRAC: das EU-Gremium zur Risikobewertung bei humanen Arzneimitteln
Der Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz der Europäischen Arzneimittelagentur ist zuständig für die Beurteilung und Überwachung von Sicherheitsaspekten für humane Arzneistoffe, und somit das maßgebliche Gremium bei der Risikobewertung. Die Aufgaben des PRAC erstrecken sich über die folgenden Bereiche, aus denen neue Erkenntnisse zu Arzneimitteln gesammelt werden und welche mögliche Quellen und Ansatzpunkte für Risikobewertungsverfahren bilden können:
- Aspekte des Risikomanagements (Risk Management Plan)
- Pharmakovigilanz-Audits (Inspektionen)
- Bewertung von Arzneimittelsicherheits-Studien nach Marktzulassung (Post authorisation safety study, PASS)
- Bewertung von Berichten über die Unbedenklichkeit von Arzneimitteln (Periodic safety update reports, PSUR)
- Erkennen und Bewerten von Risiken (Signal detection)
- Grundlegende Bewertung des Risikoprofils eines Arzneimittels oder einer Klasse von Arzneimitteln (Referral-Verfahren)
Der PRAC setzt sich aus verschiedenen Mitgliedern (Delegates) und Stellvertretern (Alternates) zusammen, die auf Grundlage ihrer persönlichen Qualifikationen in bestimmten Therapiegebieten, Erfahrung und Expertise hinsichtlich Fragen der Pharmakovigilanz ausgewählt werden:
- Je einem Vertreter und einem Stellvertreter pro Mitgliedsland der EU;
- je einem Vertreter und einem Stellvertreter aus Island und Norwegen;
- sechs unabhängigen wissenschaftlichen Experten;
- einem Mitglied und einem Stellvertreter der Gesundheitsberufe;
- einem Mitglied und einem Stellvertreter, die Patientenorganisationen repräsentieren;
- einem Vorsitzenden und einem Stellvertretenden Vorsitzenden, welche aus den Reihen der oben aufgeführten PRAC-Mitglieder gewählt werden.
Bei Bedarf holt der PRAC zusätzliche wissenschaftliche Expertise ein, ruft Expertengremien ein, fordert Fachgesellschaften zu Stellungnahmen auf oder tauscht sich mit anderen regulatorischen Behörden wie der Food and Drug Administration (FDA) in den USA aus.
Die Mitglieder des PRAC werden für drei Jahre ernannt, eine Wiederwahl ist möglich. Aus Deutschland sind Mitglieder von BfArM und PEI im PRAC vertreten. Jedes PRAC-Mitglied verfügt über eine Stimme, mit Ausnahme der Vertreter von Norwegen und Island, die kein Stimmrecht besitzen. Den Abschluss eines PRAC-Verfahrens bildet die sogenannte PRAC-Empfehlung (siehe Tab. 1). Diese Empfehlungen beruhen auf Mehrheitsentscheidungen.
Ablauf eines europäischen Risikobewertungsverfahrens
Ein Referral kann von einem Mitgliedsstaat, der Europäischen Kommission oder dem pharmazeutischen Unternehmer selbst eingeleitet werden. Den Beginn des eigentlichen Verfahrens bildet immer die sogenannte „Notification“, in der die Mitgliedsländer und betroffene pharmazeutische Unternehmer darüber in Kenntnis gesetzt werden, dass und aus welchen Gründen ein Referral-Verfahren zu einem bestimmten Arzneimittel eingeleitet wird. Im nächsten Schritt werden dann die Staaten benannt, welche bei diesem Verfahren die Federführung übernehmen, der sogenannte Rapporteur und ein oder mehrere Co-Rapporteure. Oftmals sind dies neben dem das Referral auslösende Mitgliedsland die Staaten, die auch sonst die Federführung bei der Bewertung von Risiken zu diesen Arzneimitteln haben. Der weitere Ablauf des Verfahrens erfolgt dann nach einem vorgegebenen Zeitplan (siehe Abb. 1).
Die abgebildete Zeitleiste der Abläufe während der PRAC/CHMP-Bewertung ist allerdings nur eine grobe Einteilung, denn der PRAC/CHMP kann die Zeit flexibel einteilen um auf besondere Punkte besser einzugehen und somit ein möglichst passendes Verfahren für jeden Einzelfall zu gewährleisten.
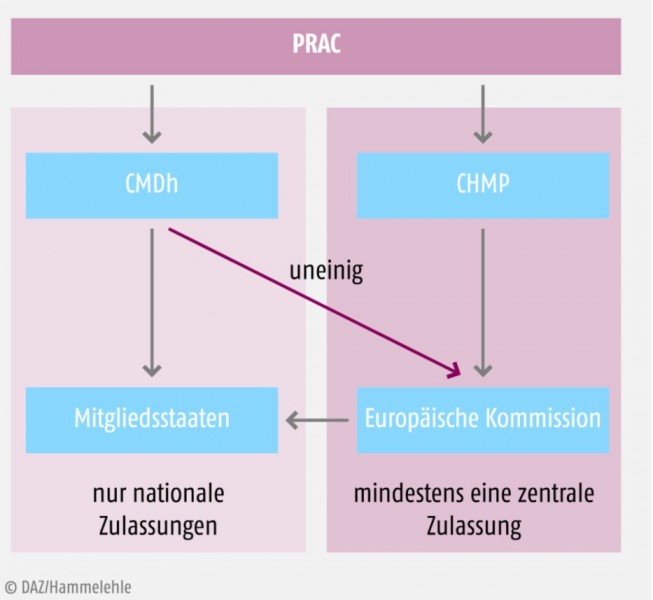
Die Rapporteure erstellen unabhängig voneinander Bewertungsberichte (Assessment Reports, AR), geben darin ihre vorläufige Einschätzung zu dem neu erkannten Risiko auf Basis der verfügbaren Daten ab und schlagen das weitere Vorgehen vor. Meist wird vor einer finalen Entscheidung eine Frageliste (List of questions, LoQ) mit der Aufforderung um Stellungnahme an die betroffenen pharmazeutischen Unternehmer zusammengestellt. Andere Länder sind in diesem Verfahren aufgerufen zu kommentieren und eigene Fragen zu stellen oder Vorschläge zur Risikominimierung zu machen. Die Koordination bei der Zusammenführung und Weiterleitung der Informationen an alle zuständigen Behörden und betroffenen pharmazeutischen Unternehmer übernimmt die EMA. Die Risikobewertung im PRAC endet mit einer Empfehlung, wie mit dem Problem weiter umzugehen ist.Die Empfehlungen des PRAC werden wiederum vom Ausschuss für Humanarzneimittel der EMA (dem CHMP) bewertet, wenn es um in der gesamten EU einheitlich zugelassene Arzneimittel geht (zentrale Zulassungen). Bei Arzneimitteln, welche nicht in der gesamten EU einheitlich zugelassen sind (nationale Zulassungen), übernimmt die Koordinierungsgruppe für Verfahren der gegenseitigen Anerkennung, im englischen Coordination Group for Mutual Recognition and Decentralised Procedures – Human (CMDh), die weitere Prüfung. Eine einstimmige Entscheidung der CMDh kann direkt an die Mitgliedsländer übermittelt werden. Nicht-einstimmige Entscheidungen des CMDh und harmonisierte Positionen des CHMP (die sogenannte opinion) werden anschließend von der Europäischen Kommission geprüft und an die Mitgliedstaaten kommuniziert (siehe Abb. 2).
Bei einigen Referrals haben die betroffenen Pharmazeutischen Unternehmer vor Verabschiedung der Entscheidung durch die Europäische Kommission die Möglichkeit, eine Neubewertung (Re-examination) zu beantragen.Das Ergebnis eines Referral-Verfahrens kann eine Änderung, ein Ruhen oder ein Widerruf der Zulassung sein. Bei einer Änderung der Zulassung bleibt das Arzneimittel mit Änderungen (bsp. Einschränkung des Anwendungsgebietes oder der Anwendungsdauer) verkehrsfähig. Das Ruhen und der Widerruf der Zulassung führen dazu, dass das betroffene Arzneimittel vom Markt genommen wird. Beim Ruhen einer Zulassung kann die Zulassung des Arzneimittels wieder erteilt werden, wenn neue Daten vorgelegt werden, die ein positives Nutzen-Risiko-Verhältnis nahelegen. Beim Widerruf einer Zulassung ist dies nicht der Fall. Mit Bekanntgabe des Widerrufs einer Zulassung darf ein Apotheker das betreffende Arzneimittel nicht mehr abgeben!



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.