














- DAZ.online
- DAZ / AZ
- DAZ 31/2012
- Hoffnung Gentherapie
Gentherapie
Hoffnung Gentherapie
Adrenoleukodystrophie (X-ALD)
Die Adrenoleukodystrophie ist eine X-chromosomal rezessiv vererbte Erkrankung (X-ALD), die erstmals 1923 erkannt wurde und durch eine Mutation im Gen ABCD1 hervorgerufen wird. ABCD1 codiert für das Adrenoleukodystrophie-Protein (ALDP). Dieses Protein befindet sich in der Membran der Peroxisomen und gehört zu den Transportproteinen, die eine ATP-bindende Kassette aufweisen. Infolge bestimmter Mutationen können gesättigte sehr langkettige Fettsäuren (very long chain fatty acids, VLCFA) nicht mehr richtig in Membranen der Peroxisomen abgebaut werden (Abb. 1), sodass sie in den Zellen akkumulieren. Dadurch verändern sich die physiologischen Eigenschaften der Biomembranen. Beispielsweise hat man bei ALD-Patienten festgestellt, dass die Viskosität der Erythrozytenmembran erhöht ist. Bei den Nervenzellen wird vermutet, dass die gesättigten VLCFA die Stabilität der Myelinmembran beeinträchtigen und dadurch eventuell deren Abbau fördern. Allerdings sind hier auch Entzündungsprozesse beteiligt.
Zur Diagnose der ALD werden neben der Untersuchung der Seh- und Hörfähigkeit vor allem die Konzentration gesättigter VLCFA im Plasma herangezogen. Drei Testparameter werden bestimmt:
- die Konzentration der Cerotinsäure (C26:0),
- das Verhältnis von Lignocerinsäure zu Behensäure (C24:0/C22:0) und
- das Verhältnis von Cerotinsäure zu Behensäure (C26:0/C22:0).
Alle drei Parameter sind bei ALD-Patienten deutlich erhöht.
Da die Erkrankung X-chromosomal vererbt wird, sind üblicherweise Männer betroffen, während Frauen die Mutation weitergeben. Aber auch ca. 50% der heterozygoten Trägerinnen leiden im mittleren oder höheren Alter trotz einer vorhandenen intakten Genkopie an neurologischen Defekten. Die Inzidenz der Erkrankung wird auf 1:17.000 Neugeborenen geschätzt, wobei alle Ethnien gleichmäßig betroffen sind.
Formen der X-ALD
Die Adrenoleukodystrophie kann in verschiedenen Formen auftreten. Die klassische kindliche Form ist die schwerste Variante. Sie tritt bei Jungen im Alter von 4 bis 10 Jahren auf, und auf sie entfallen ungefähr 35% der Erkrankungen. Erste Symptome können Konzentrationsstörungen, Lernschwierigkeiten und Wesensveränderungen sein. Im weiteren Verlauf kommt es zu Einschränkungen der Sehkraft und des Hörvermögens sowie der Bewegungskoordination. Der Tod tritt meist zwei bis vier Jahre nach dem Auftreten der ersten Symptome ein.
Die häufigste Form der X-ALD ist die Adrenomyeloneuropathie. Betroffene Patienten verspüren die Symptome, die aus Schäden im Rückenmark und der peripheren Nervenzellen resultieren, im zweiten bis dritten Lebensjahrzehnt. Es kommt zur Beeinträchtigung der Gehfähigkeit, sodass die Betroffenen nach einiger Zeit einen Rollstuhl brauchen. Bei ca. 20% dieser Patienten findet außerdem ein zerebraler Myelinabbau statt, was ihre Lebenserwartung sehr stark verringert.
Wegen der Schädigung der Nebennierenrinde sind ALD-Patienten auf eine Ersatztherapie mit Nebennierenrindenhormonen angewiesen. Daneben werden diätetische Maßnahmen propagiert, wie z. B. „Lorenzos Öl“. Die schweren Fälle müssen mit Stammzelltherapie oder neuerdings auch mit einer Gentherapie behandelt werden.
Lorenzos Öl
1992 wurde die Geschichte von Lorenzo Odone und seinen Eltern verfilmt. Der 1978 auf den Komoren geborene Junge zeigte im Alter von sechs Jahren die ersten Symptome einer X-ALD. Mit der Aussicht konfrontiert, dass ihr Sohn wahrscheinlich noch maximal zwei Jahre zu leben habe, suchten seine Eltern nach möglichen Therapien. So mischten sie verschiedene Oliven- und Rapsöle, um sie dem Kind zu verabreichen. Es resultierte ein Spezialöl, das unter der Bezeichnung „Lorenzos Öl“ sogar patentiert wurde. Dieses Öl ist eine 1:4-Mischung der Triglyceride Glyceroltrierucat (GTE) und Glyceroltrioleat (GTO), die die einfach ungesättigten Fettsäuren Erucasäure (C22:1 ω-13) bzw. Ölsäure (C18:1 ω-9) enthalten. Mithilfe dieser Diät wurde Lorenzo Odone 30 Jahre alt.
Die Ratio der Anwendung liegt darin, die Synthese der gesättigten Fettsäuren Lignocerinsäure (C24:0) und Cerotinsäure (C26:0) aus Behensäure (C22:0) durch die Erucasäure und Ölsäure kompetitiv zu hemmen. Durch die Einnahme von Lorenzos Öl können tatsächlich die VLCFA-Plasmawerte bei X‑ALD-Patienten innerhalb von vier Wochen normalisiert werden. Allerdings kann die Einnahme des Öls u. a. zu Thrombo- und Leukozytopenien, erhöhten Leberenzymwerten und Kardiomyopathien führen. Seit etlichen Jahren wird kontrovers diskutiert, ob das Öl letztlich einen klinischen Effekt bei X-ALD-Patienten hat.
Erucasäure scheint zwar die Blut-Hirn-Schranke zu überwinden, allerdings kann sie wohl eine bereits begonnene Demyelinisierung der Nervenzellen nicht mehr stoppen, geschweige denn rückgängig machen. Eventuell könnte das Öl jedoch denjenigen X-ALD-Patienten helfen, die noch keine Schädigungen im Gehirn aufweisen.
Während das Hessische Landessozialgericht 2007 Lorenzos Öl die Erstattungsfähigkeit zugesprochen hatte, hob das Bundessozialgericht in Kassel diesen Beschluss am 28. Februar 2008 auf, sodass die monatlichen Kosten von ca. 700 Euro nicht mehr von den gesetzlichen Krankenkassen übernommen werden. Das Bundessozialgericht begründete die Entscheidung damit, dass Lorenzos Öl die Zulassung als Arzneimittel fehlt und deshalb nur als Lebensmittel eingestuft werden kann, das nicht durch die Krankenkasse bezahlt wird.
Die ALD-Gen-/Stammzelltherapie in Paris
Die übliche Therapie der schweren X-ALD ist die allogene Transplantation hämatopoetischer Stammzellen eines Spenders. Diese Zellen enthalten natürlich das intakte Gen ABCD1 und synthetisieren das physiologische Protein, sodass der Defekt kompensiert wird, falls die Zellen das richtige Kompartiment erreichen. Sofern die zerebrale Schädigung noch nicht weit fortgeschritten ist, können gesunde myelomonozytische Zellen, die sich aus den transplantierten Stammzellen gebildet haben, in das Gehirn des ALD-Patienten einwandern und dort einen Teil der Mikrogliazellen ersetzen. Dadurch wird im besten Fall eine weitere Demyelinisierung der Nervenzellen verhindert. Sind allerdings keine passenden Knochenmarkspender verfügbar, fällt diese Therapieoption weg.
Genau aus diesem Grund wurde 2005 in Paris beantragt, zwei Jungen im Alter von 7 und 7,5 Jahren, für die es keine passenden Knochenmarkspender gab, mit eigenen gentechnisch modifizierten hämatopoetischen Stammzellen zu behandeln. Bei beiden Jungen war bereits jeweils ein älterer Bruder an der schweren Verlaufsform der X-ALD verstorben. Die Idee war nun, bei den Kindern über die Verabreichung von Granulozyten-Kolonie-stimulierendem Wachstumsfaktor (G-CSF) hämatopoetische Stammzellen (HSC) aus dem Knochenmark zu mobilisieren und diese Zellen aus dem Blut zu isolieren. Anschließend sollte in die hämatopoetischen Stammzellen das intakte Gen ABCD1 integriert werden. Die so modifizierten Stammzellen sollten dann den Kindern reinfundiert werden. Damit wäre quasi die allogene Knochenmarktransplantation simuliert, und im Idealfall ließe sich auch die Demyelinisierung der Nervenzellen aufhalten.
Somit wurden zwei Techniken kombiniert, die bisher nur bedingt erfolgreich als Therapien eingesetzt worden waren: eine Stammzelltherapie und eine Gentherapie.
Die Genfähre: ein lentiviraler HIV-1-Abkömmling
Noch mehr mag es verwundern, dass für den Gentransfer bei den X-ALD-Patienten ein Vektor verwendet wurde, der sich vom HIV-1 ableitet. Wie kann es sein, dass ein potenziell derart gefährliches Virus als Therapeutikum bei Kindern eingesetzt wird?
Nach vielen Versuchen mit unterschiedlichsten Transfersystemen ist man mittlerweile bei den Lentiviren angekommen, was letztlich den Anforderungen an eine erfolgreiche Gentherapie geschuldet ist:
ein leicht zu veränderndes, möglichst sicheres Vektorsystem, das idealerweise beliebig große DNA-Abschnitte transportieren kann,
eine stabile Integration der DNA in das Genom der Zelle und folglich eine nachhaltige Expression des eingebrachten Gens und die Weitergabe des Gens bei der Zellteilung und
die Möglichkeit, sowohl sich teilende als auch sich nicht teilende Zellen in möglichst unterschiedlichen Gewebetypen zu erreichen und zu modifizieren.
Natürlich lässt sich als Vektor nicht einfach die unmodifizierte HIV-1-RNA verwenden. Vielmehr wird ein Vektorsystem eingesetzt, bei dem es sich um eine ausgeklügelte Kombination von unterschiedlichen Vektoren mit geeigneten Verpackungszelllinien handelt; die Anwendung dieser Genfähre ist extrem sicher.
Das Vektorsystem
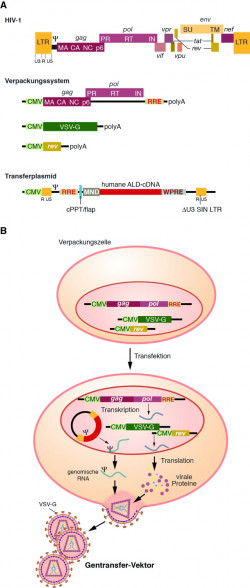
Das eigentliche Transferplasmid enthält neben der cDNA des Transgens nur noch diejenigen Bereiche von HIV-1, die absolut notwendig sind, um die RNA in ein Viruspartikel zu verpacken (das Verpackungssignal Ψ), diese in der Zielzelle revers zu transkribieren und die resultierende cDNA in das Genom der Zelle zu integrieren (die long terminal repeats, LTR und das Rev-responsive element, RRE). Somit trägt das Transferplasmid ausschließlich virale Kontrollelemente aber keinerlei Information für virale Proteine.
Da das Einbringen des nackten Transferplasmids in eine Zielzelle sehr viel ineffizienter erfolgt als die Infektion mit einem Viruspartikel, wird die genetische Information mit einem Trick in ein Virion verpackt. Dazu wird eine Verpackungszelllinie verwendet, die ihrerseits bereits Teile des HIV-1-Genoms in Form von Verpackungsplasmiden enthält (Abb. 2A). Eines dieser Plasmide trägt die Information für die HIV-Core-Proteine Gag und Pol, sodass in der Verpackungszelllinie die viralen Proteine Protease (PR), reverse Transkriptase (RT) und Integrase (IN) bereitgestellt werden.
Anstelle der viralen Env-Proteine, die beim HI-Virion durch die Oberflächen-Glykoproteine gp120 (surface subunit, SU) und gp41 (transmembrane, TM) repräsentiert sind, wird in der Verpackungszelllinie das Oberflächen-Glykoprotein G des vesikulären Stomatitis-Virus (VSV-G) exprimiert. Dadurch wird der Tropismus, d. h. die Infektionsspezifität des gebildeten Virions deutlich erhöht. Während HI-Viren über die Interaktion zwischen gp120 und CD4 nur T-Zellen infizieren können, kann das so modifizierte Virion nun über das VSV-G-Glykoprotein an ein deutlich breiteres Spektrum von Zielzellen andocken.
Als weitere HI-virale Informationseinheit wird noch das Rev-Protein in der Verpackungszelllinie exprimiert, das im Zellkern vorliegt und dort dafür sorgt, dass alle mRNAs, die eine RRE-Sequenz tragen, effizient ins Zytoplasma transportiert werden. Alle Fremd-DNAs in der Verpackungszelllinie werden unter der Kontrolle eines Cytomegalievirus-Promotors (CMV) transkribiert.
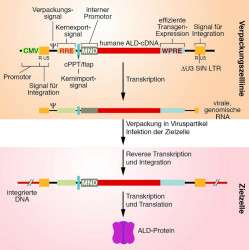
Die RNA des Transferplasmids wird nun als virales Genom mit den nötigen viralen Proteinen in Viruspartikel verpackt. Diese Partikel können aus dem Kulturüberstand der Verpackungszelllinie isoliert und zur Infektion der Zielzelle eingesetzt werden (Abb. 2B). Sobald das Virus in die Zelle aufgenommen wurde, wird die virale genomische RNA revers transkribiert und in das Genom der Zielzelle integriert. Der interne MND-Promotor (myeloproliferativer Sarcomavirus-Promotor) sorgt für eine effiziente und stabile Transkription der therapeutisch relevanten ALD-cDNA (Abb. 3).
Die Zielzellen: hämatopoetische Stammzellen
Ganz analog wie bei der Vorbereitung einer allogenen Knochenmarktransplantation wurden auch den ALD-Patienten nach Stimulation mit Granulozyten-Kolonie-stimulierendem Wachstumsfaktor (G-CSF) mononukleäre Zellen des peripheren Blutes (peripheral blood mononuclear cells, PBMCs) entnommen. Aus dieser Zellpopulation wurden diejenigen Zellen isoliert, die auf ihrer Oberfläche CD34 exprimieren. CD34 ist derzeit „das“ Identifikationsmerkmal für hämatopoetische Stammzellen (Abb. 4). Allerdings weiß man inzwischen, dass nur ca. 5 bis 20% dieser CD34+-Zellen echte hämatopoetische Stammzellen (HSC) sind. Bei dem Rest handelt es sich um Vorläuferzellen und weiße Blutzellen in verschiedenen Differenzierungsstadien.
Der Vorteil, HSC aus peripherem Blut zu isolieren, liegt nicht nur in der leichteren Verfügbarkeit der Zellen, sondern auch darin, dass diese Stammzellen leichter Fremdgene aufnehmen und schneller anwachsen als diejenigen Stammzellen, die direkt aus dem Knochenmark isoliert wurden.
Man hat berechnet, dass jede Stammzelle 17 bis 19,5 Zellteilungen durchläuft, um schließlich zu einer Blutzelle zu differenzieren. Das entspricht einem Amplifikationsfaktor von ca. 130.000 bis 720.000! Wiederholt wurde mittlerweile auch gezeigt, dass HSCs in verschiedenste Gewebetypen wie Gehirn, Muskel und Leber differenzieren können und diese so regenerieren. Daher erscheint es plausibel, dass auch gentechnisch modifizierte Mikroglia-Vorläuferzellen aus dem Blut über die Blut-Hirn-Schranke in das Gehirn einwandern, um dort die Entzündungsreaktion bei den ALD-Patienten zu modulieren.
An der französische Gentherapiestudie nahmen als erste Patienten zwei Jungen im Alter von 7 bzw. 7,5 Jahren teil, die bereits eine fortgeschrittene zerebrale, neuroinflammatorische Demyelinisierung zeigten. Außerdem hatten beide einen Bruder, der an zerebraler X‑ALD verstorben war. Insofern war die Prognose für die beiden Jungen als extrem schlecht einzustufen. Da auch kein geeigneter Knochenmarkspender zur Verfügung stand, waren alle Voraussetzungen gegeben, mit diesen beiden Patienten diese riskante Intervention auch ethisch zu vertreten.
Von beiden Patienten wurden nach G-CSF-Stimulation periphere Blutzellen gewonnen und unter ihnen die HSC angereichert. Für den Gentransfer wurden die Zellen mit Virionen, die aus der geschilderten Verpackungsstrategie gewonnen wurden, mit einem Multiplikationsfaktor (multiplicity of infection, MOI) von 25 infiziert. Damit sollte erreicht werden, dass ein bis zwei Kopien der revers transkribierten transgenen RNA in das Genom der Zielzelle integrieren. 5% der behandelten Zellen wurden abgezweigt, um zum einen zu zeigen, dass keine Replikations-kompetenten Lentiviren (RCL) gebildet werden. Zum anderen wurde überprüft, ob in den infizierten HSCs das Transgen auch tatsächlich exprimiert wird. Nachdem diese Tests erfolgreich verlaufen waren, wurden bei den Jungen zunächst die verbliebenen Knochenmarkzellen durch die Gabe von Cyclophosphamid und Busulfan zerstört. Danach wurden ihnen mehr als 4,6 × 106 gentechnisch modifizierte hämatopoetische Stammzellen/kg Körpergewicht reinfundiert.
Auch fünf Jahre nach der Therapie lässt sich bei den beiden Patienten noch bei 10 bis 13% der PBMCs das integrierte Transgen nachweisen. Und erfreulicherweise ist die Demyelinisierung nicht weiter fortgeschritten.
Mittlerweile sind noch zwei weitere Kinder analog behandelt worden, von denen jedoch eines einen Rückfall erlitt.
Stammzellen als neue Therapieoptionen?
Stammzellen sind wichtige Zellen im lebenden Organismus: Sie sind undifferenziert und können sich durch Teilung vermehren oder unter bestimmten Bedingungen zu gewebespezifischen Zellen differenzieren. Dadurch sind sie in gewissem Umfang in der Lage, geschädigtes Gewebe zu reparieren.
Nachdem bereits 1981 embryonale Stammzellen aus der Maus isoliert wurden, konnte man 1998 auch entsprechende Zellen aus menschlichen Embryonen, die für die In-vitro-Fertilisation kultiviert worden waren und nicht mehr benötigt wurden, gewinnen. Neben den embryonalen Stammzellen, die aus den inneren Zellen der Blastozyste, also einem drei bis fünf Tage alten Embryo, isoliert werden, kommen im Körper auch adulte Stammzellen vor, die z. B. im Knochenmark oder im Gastrointestinaltrakt für die regelmäßige Zellerneuerung zuständig sind.
2006 konnten Kulturbedingungen gefunden werden, mit denen sich bereits differenzierte Zellen, wie beispielsweise Haut-Fibroblasten, wieder in einen Stammzell-ähnlichen Zustand zurückversetzen lassen. Diese auch „induzierte pluripotente Stammzellen“ (iPSC) genannten Zellen könnten im Rahmen einer regenerativen Therapie bei verschiedenen Erkrankungen wie Diabetes, Morbus Parkinson oder auch Adrenoleukodystrophie eingesetzt werden. Ihre großen Vorteile liegen darin,
dass sie – anders als embryonale Stammzellen – ohne ethische Bedenken gewonnen werden können,
dass sie vom Patienten selbst stammen, sodass das Problem der Abstoßung von Fremdgewebe hier nicht auftritt, und
dass sie leicht herzustellen sind, während adulte Stammzellen in den Geweben in nur geringer Zahl vorkommen und – außer im Knochenmark – schwer zu finden und zu isolieren sind.
In Maus- und Rattenmodellen konnten durch iPSC, die aus Haut-Fibroblasten der Tiere gewonnen wurden, dopaminerge Neuronen für Parkinson-Ratten kultiviert und anschließend transplantiert werden. Ähnlich erfolgreich war beispielsweise eine Transplantation von Faktor-VIII-produzierenden Endothelzellen in Hämophilie-A-Mäuse oder von Insulin-produzierenden Pankreaszellen in Typ-1-diabetische Mäuse. Allerdings muss die Differenzierung der iPSC zu Neuronen oder Endothelzellen sehr sorgfältig durchgeführt werden. Bleibt auch nur eine undifferenzierte iPSC übrig, besteht die große Gefahr, dass sie zu einem Tumor auswächst.
Einen großen Nachteil scheinen iPSCs aber auch zu besitzen: Ihr Genom scheint deutlich instabiler zu sein als das von embryonalen Stammzellen. Daher kann es zu einer Anhäufung von Mutationen kommen, die dann auch eine Entartung der Zellen induzieren können. Aus diesem Grund wird mittlerweile versucht, Fibroblasten auf direktem Weg umzuprogrammieren, beispielsweise zu induzierten dopaminergen Neuronen (iDAN). Einige klinische Phase-I-Studien werden bereits durchgeführt, bei denen sowohl embryonale als auch induzierte pluripotente Stammzellen oder aber genetisch korrigierte iPSC, in die beispielsweise ein intaktes Gen für α1-Antitrypsin eingebracht wurde, eingesetzt werden.
Es bleibt abzuwarten, wie sich diese Therapien bewähren und welche Probleme dabei noch auftreten. Wichtig wird sein, die beteiligten Patienten genau zu beobachten und den Therapieverlauf sorgfältig zu dokumentieren. Nur so können der therapeutische Nutzen und die Risiken bestimmt und gegeneinander abgewogen werden.
InternetDatenbank der X-Chromosom-gekoppelten Adrenoleukodystrophie: www.x-ald.nl |
Literatur Ärzte Zeitung 31 (2012), 108. Kemp S, Berger J, Aubourg P. X-linked adrenoleukodystrophy: Clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta (2012); doi:10.1016/j.bbadis.2012.03.012. Bundessozialgericht, Urteil vom 28.2.2008, B 1 KR 16/07 R. Cartier N, et al. Lentiviral Hematopoietic Cell Gene Therapy for X-Linked Adrenoleukodystrophy. Methods Enzymol 507 (2012), 187 – 198. Berger J, et al. Current and Future Pharmacological Treatment Strategies in X-Linked Adrenoleukodystrophy. Brain Pathol 20 (2010), 845 – 856. Sandhaus R.A. Gene Therapy Meets Stem Cells. N Engl J Med 366 (2012), 567 – 569. Biffi A, Aubourg P, Cartier N. Gene therapy for leukodystrophies. Hum Mol Gen 20 (2011), R42 – R53. Mummery C. Induced Pluripotent Stem Cells – A Cautionary Note. N Engl J Med 364 (2011), 2160 – 2162. Sakuma T, Barry MA, Ikeda Y. Lentiviral vectors: basic to translational. Biochem J 443 (2012), 603 – 618. Cappa M, et al. A mixture of oleic, erucic and conjugated linoleic acids modulates cerebrospinal fluid inflammatory markers and improve somatosensorial evoked potential in X-linked adrenoleukodystrophy female carriers. J Inherit Metab Dis (2011); DOI 10.1007/s10545-011-9432-3. Moser HW, et al. "Lorenzo’s Oil" Therapy for X-linked Adrenoleukodystrophy: Rationale and Current Assessment of Efficacy. J Mol Neurosci 33 (2007), 105 – 113. Cartier N, et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy. Science 326 (2009), 818 – 823. Naldini L. A Comeback for Gene Therapy. Science 326 (2009), 805 – 806. Cha H-J, Hwang ES. Current Status of Biology, Bioengineering, and Therapeutic Potential of Stem Cells. Arch Pharm Res 35 (2012), 193 – 196. Grabel L. Prospects for Pluripotent Stem Cell Therapies: Into the Clinic and Back to the Bench. J Cell Biochem 1 (2012), 381 – 387.
Autoren
Prof. Dr. Theo Dingermann, Dr. Ilse Zündorf, Institut für Pharmazeutische Biologie, Max-von-Laue-Str. 9, 60438 Frankfurt



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.