













- DAZ.online
- DAZ / AZ
- DAZ 45/2010
- Regenerative Medizin
Recht
Regenerative Medizin
Gentherapie
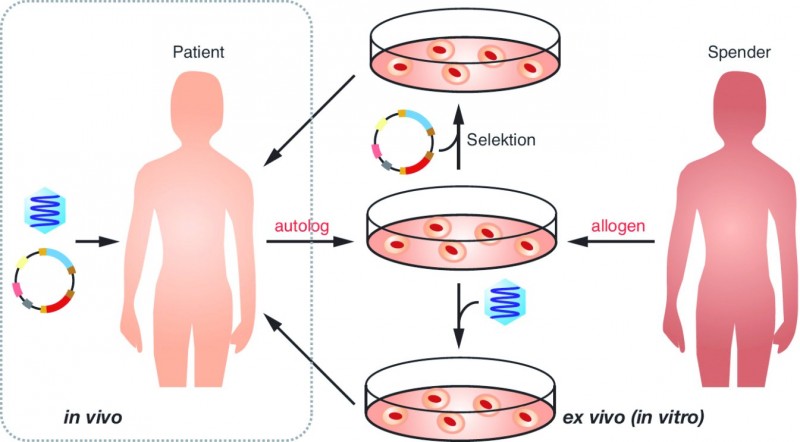
Die therapeutische DNA oder RNA kann dem Patienten "nackt" oder mithilfe eines Vektors (z. B. Adenovirus) verabreicht werden (links). Eine andere Option ist die
Applikation der DNA oder RNA in genetisch modifizierten Zellen, wobei die Zellen von dem Patienten selbst (autolog) oder von einem Spender stammen können (allogen).
Grafik aus Dingermann et al., Gentechnik – Biotechnik [11].
Gewebespenden in der regenerativen Medizin
Die Verwendung von Gewebespenden zur Wiederherstellung von Körperfunktionen ist seit Langem etabliert. Die erste erfolgreiche Hornhauttransplantation wurde 1905 vom österreichischen Augenarzt Eduard Konrad Zirn vorgenommen, das Transplantat blieb über ein Jahr lang transparent. 1964 wurde erstmals eine Herzklappe vom Schwein in einen Menschen verpflanzt. Bis zum heutigen Tag wurden in verschiedensten Indikationen unzählige Gewebetransplantationen durchgeführt (Tab. 1). Um den großen Bedarf an Gewebespenden zu decken, bildeten sich nationale und internationale Netzwerke, sogenannte Gewebebanken. Zugleich stellten sich regulatorische Fragen:
Wie kann eine möglichst optimale Versorgung für alle Bürger sichergestellt werden ohne Beschränkungen an nationalen Grenzen?
Wie kann die Unbedenklichkeit der Gewebespenden gewährleistet werden?
Der Punkt der Unbedenklichkeit umfasst so unterschiedliche Bereiche wie Freiheit von mikrobiellen Erregern – dabei spielt auch die regional unterschiedliche Verbreitung von Krankheitserregern eine Rolle – , die Haltbarkeit der Gewebespenden bei Lagerung und Transport, die Pharmakovigilanz und die Rückverfolgbarkeit des Produktes für den Fall, dass zu einem späteren Zeitpunkt bei dem Spender oder einem (von mehreren) Empfänger Komplikationen auftreten, die durch die Gewebespende auch andere Empfänger betreffen können, z. B. aufgrund einer nicht identifizierten Infektion des Spenders.
| Tab. 1: Anzahl der jährlichen Gewebetransplantationen in Deutschland und weltweit | ||
| Gewebeart | Anzahl der Transplantationen pro Jahr* | |
| in Deutschland | weltweit | |
| Hornhaut | 4.500 – 6.000 | 100.000 |
| Herzklappen | 15.000 | 250.000 |
Knochenmark – autolog – allogen | 75.000 30.000 | ? USA: 900.000 |
* Quelle: www.gewebenetzwerk.de/gewebearten/herzklappen.html;
über die Transplantation von Haut und Blutgefäßen sind keine Angaben verfügbar
** Quelle: www.bag.admin.ch/
transplantation/00697/01680/01685/index.html?lang=de
Drohende Gefahren
Dass es sich dabei um ein realistisches Szenario handelt, zeigen u. a. zwei Fälle von Tollwut aufgrund von Organtransplantationen, einer in den USA im Jahr 2004 und der andere in Deutschland 2005. Bei dem letzteren Fall war die Organspenderin im Oktober 2004 durch einen Hundebiss in Indien infiziert worden. Als die Frau im Dezember an einem Herzstillstand verstarb, waren keine Symptome bekannt, die auf diese Infektion hingewiesen hätten. Von den sechs Patienten, welche Organe oder Gewebe der Spenderin erhalten hatten, verstarben drei an Tollwut. Ein Patient, der die Leber der Frau erhalten hatte, war früher gegen Tollwut geimpft worden und so gegen die Krankheit geschützt. Die beiden Patienten, die die Corneae der Spenderin erhalten hatten, erkrankten ebenfalls nicht. Da es dokumentierte Fälle von Tollwutübertragung durch Cornea-Transplantation gibt, wurden die Corneae bei beiden Empfängern ausgetauscht und auf Rabiesviren untersucht – glücklicherweise mit negativem Ergebnis.
Geweberichtlinien der EG
Um die Unbedenklichkeit der Gewebespenden zu gewährleisten, haben das Europäische Parlament und der Europäische Rat mehrere Richtlinien verabschiedet:
Die Richtlinie 2004/23/EG, auch Geweberichtlinie genannt, legt als generellen Rahmen die "Qualitäts- und Sicherheitsstandards für die Spende, Beschaffung, Testung, Verarbeitung, Konservierung, Lagerung und Verteilung von menschlichem Gewebe" fest [1].
Die Richtlinie 2006/86/EG regelt die Anforderungen an Herstellung, Verteilung, Zwischenfallmeldungen und die Rückverfolgbarkeit bei Geweben und Zellen [2].
Die Richtlinie 2006/17/EG umfasst technische Vorschriften für die Spende, Beschaffung und Testung von menschlichen Geweben und Zellen [3].
Diese Regelungen sollen sicherstellen, dass die hier verwendeten Gewebespenden überall nach den gleichen strengen Standards entnommen, getestet und gelagert werden. Allerdings müssen EG-Richtlinien von jedem Mitgliedstaat der Europäischen Gemeinschaft in nationales Recht umgesetzt werden. Da dieses mit unterschiedlicher Geschwindigkeit geschieht und die nationalstaatlichen Regierungen einen gewissen Ermessensspielraum haben, ist eine vollkommen einheitliche Regelung in Europa zurzeit noch nicht gegeben und wird unter Umständen noch der weiteren Steuerung durch die EG bedürfen. Einzelheiten zur Implementierung der Gesetzgebung zu Geweben und Zellen in den einzelnen Mitgliedstaaten finden sich in dem Bericht "Summary Table of Responses from Competent Authorities for Tissues and Cells: Questionnaire on the transposition and implementation of the European Tissues and Cells regulatory framework” [4].
In Deutschland wurden die genannten Richtlinien im Gewebegesetz umgesetzt [5]. Damit wurden Gewebeprodukte bzw. Zubereitungen, die aus Geweben bestehen oder daraus hergestellt worden sind, als Arzneimittel in das Arzneimittelgesetz aufgenommen.
Gewebe sind einzelne Zellen und alle aus Zellen bestehenden Bestandteile des menschlichen Körpers zu verstehen, die keine Organe sind.
Gewebeprodukte (= Gewebezubereitungen) sind beispielsweise Augenhornhaut (Cornea), Bindegewebe (Faszien), embryonale und fötale Gewebe, Herzgefäße, Herzklappen, Knochenmark, Knochenpräparationen, aber auch einzelne Zellen, z. B. Stammzellen [6].
Menschliche Samen- und Eizellen einschließlich Keimzellen (imprägnierte, d. h. befruchtete Eizellen vor der Kernverschmelzung) und Embryonen sind weder Arzneimittel noch Gewebezubereitungen (vgl. § 4 Abs. 30 AMG i.V.m. § 1a Nr. 4 TPG [7]).
Das Gewebegesetz regelt auch solche Gewebezubereitungen, die mit nicht-industriellen Verfahren, die in der Europäischen Union hinreichend bekannt sind, be- oder verarbeitet werden und deren Wirkungen und Nebenwirkungen aus dem wissenschaftlichen Erkenntnismaterial ersichtlich sind. Dies betrifft insbesondere Gewebebanken in Krankenhäusern, die vorher nicht der Überwachung unterlagen, weil beispielsweise nur geringe Zahlen an Gewebezubereitungen mit wenig Aufwand für den Eigenbedarf hergestellt und vorgehalten wurden. Medizinische Einrichtungen oder Krankenhausabteilungen, die solche Gewebezubereitungen be- und verarbeiten (entnehmen, verarbeiten, lagern, prüfen oder Inverkehrbringen), bedürfen dafür nun einer Erlaubnis durch die zuständige Landesbehörde. Darüber hinaus benötigen sie für das Inverkehrbringen der Gewebezubereitungen eine Genehmigung durch das Paul-Ehrlich-Institut und sind verpflichtet, die Anzahl der hergestellten, gelagerten und verwendeten Gewebezubereitungen diesem Institut zu melden.
An die Erlaubnis zur Be- und Verarbeitung sind Anforderungen an das Qualitätsmanagement, die Rückverfolgbarkeit von Spendern und Empfängern über einen Zeitraum von 30 Jahren, und die Pharmakovigilanz geknüpft, sodass bei auftretenden Komplikationen alle möglicherweise betroffenen Empfänger identifiziert und entsprechend behandelt werden können. Außerdem lässt sich durch die Meldepflicht der Umfang der ver- und bearbeiteten Gewebezubereitungen in Deutschland erfassen.
Arzneimittel für neuartige Therapien (ATMP)
Neue wissenschaftliche Fortschritte in der Zell- und Molekularbiotechnologie haben zur Entwicklung von Arzneimitteln für neuartige Therapien (Advanced Therapy Medicinal Products, ATMP) geführt (Tab. 2). Dazu gehören neben den Gentherapeutika die somatischen Zelltherapeutika und die biotechnologisch bearbeiteten Gewebeprodukte (Tissue-Engineered Products). Während der Wirkstoff eines Gentherapeutikums grundsätzlich eine rekombinante (also biotechnologisch erzeugte) Nucleinsäure ist [9], sind die beiden anderen Produktgruppen zellbasiert; dabei wurden die Zellen entweder substanziell verändert (z. B. in vitro vermehrte und anschließend auf einer biodegradierbaren Membran gezüchtete Keratinozyten als Hautersatz), oder sie dienen im Empfänger im Wesentlichen nicht derselben / denselben Funktion(en) wie im Spender (z. B. hämatopoetische Stammzellen zur Myokard-Regeneration) [9]. Allerdings unterscheiden sich die beiden Produktgruppen durch ihre Anwendung: Die somatische Zelltherapie dient zur Diagnose, Vorbeugung oder Behandlung von Krankheiten. Werden dem Produkt jedoch Eigenschaften zur Regeneration, Wiederherstellung oder zum Ersatz menschlichen Gewebes zugeschrieben, handelt es sich um ein biotechnologisch bearbeitetes Gewebeprodukt.
Während die Gentherapie und die somatische Zelltherapie bereits vorher als ATMP definiert waren, wurden die biotechnologisch bearbeiteten Gewebeprodukte erstmals in der Verordnung (EG) Nr. 1394/2007 (ATMP-Verordnung) definiert [10]. Darin ist festgelegt, dass ATMP in der EG nur von der Europäischen Arzneimittelbehörde EMA zugelassen werden dürfen (zentralisiertes Zulassungsverfahren, s. u.).
Gentherapeutika
Der Wirkstoff eines Gentherapeutikums ist eine rekombinante Nucleinsäuresequenz (RNA oder DNA). Dabei kann der Wirkstoff allein oder mithilfe eines Virus oder in einer genetisch modifizierten Zelle verabreicht werden (Abb. Gentherapie) [11].
Ein Beispiel für Gentherapie ist die Therapie einer Form der schweren kombinierten Immundefizienz (Severe combined immunodeficiency, SCID), die Ende der 90er Jahre in Frankreich an insgesamt zehn Kindern durchgeführt wurde. Bei Patienten mit SCID ist die Differenzierung der Blutzellen aus den Blutstammzellen derart gestört, dass es zu einer Fehlfunktion oder einem Mangel an Lymphozyten kommt und die zelluläre Immunantwort mangelhaft ist. Wegen der stark erhöhten Infektionsanfälligkeit beträgt die Lebenserwartung der Patienten nur Monate bis einige Jahre. Die einzige Therapieoption ist eine HLA-kompatible allogene Knochenmarktransplantation. Bei der Form X‑SCID tritt ein Defekt der γC-Untereinheit verschiedener Zytokinrezeptoren auf, was eine korrekte Reifung der Lymphozyten behindert; für den Defekt ist ein mutiertes Gen verantwortlich, das auf dem X-Chromosom liegt. (Die X-SCID betrifft hauptsächlich Jungen, weil diese nur ein X-Chromosom haben, während Mädchen mit dem "normalen" Gen auf ihrem zweiten X-Chromosom die Mutation kompensieren können.)
Durch den Einbau des "normalen" Gens in Blutstammzellen lässt sich die Entwicklung der Lymphozyten und damit die Ausbildung eines funktionalen Immunsystems wiederherstellen. Als Vektor für den Gentransfer diente ein Retrovirus, das in die Blutstammzellen eindringt, ohne sich weiter zu vermehren und die Zellen zu zerstören. Die so in das Patientengenom integrierte gesunde Form des Interleukinrezeptors γ ermöglichte die Ausreifung der Blutstammzellen zu funktionalen Lymphozyten und führte zur erwarteten Heilung. Allerdings trat als schwere Nebenwirkung in zwei Fällen eine akute Leukämie auf. Diese wurde direkt durch den eingeschleusten Vektor ausgelöst, vermutlich durch die Überexpression eines Onkogens am Integrationsort im Genom.
Somatische Zelltherapeutika
Ein somatisches Zelltherapeutikum ist ein biologisches Arzneimittel,
dessen Zellen oder Gewebe substanziell bearbeitet wurden, sodass biologische Merkmale, physiologische Funktionen oder strukturelle Eigenschaften, die für die beabsichtigte klinische Verwendung relevant sind, verändert wurden, oder
dessen Zellen oder Gewebe im Empfänger im Wesentlichen nicht derselben / denselben Funktion(en) dienen wie im Spender (nicht-homologe Verwendung) oder
das zum Teil aus substanziell manipulierten oder nicht-homolog verwendeten Zellen besteht.
Ein Beispiel sind die sogenannten Tumorvakzinen. Diese bestehen aus körpereigenen (autologen) Immunzellen, die ex vivo mit Proteinen in Kontakt gebracht werden, welche spezifisch für den Tumor sind. Dadurch werden die Immunzellen angeregt, die Tumorzellen zu vernichten. Eine Tumorvakzine ist das in den USA am 29. April 2010 zur Behandlung des Prostatakarzinoms zugelassene Arzneimittel Provenge™ (Sipuleucel-T) [12].
Biotechnologisch bearbeitete Gewebeprodukte
Wie somatische Zelltherapeutika bestehen auch biotechnologisch bearbeitete Gewebeprodukte ganz oder teilweise aus Geweben und Zellen, die substanziell verändert sind oder nicht-homolog verwendet werden doch unterscheiden sie sich durch ihre Wirkweise. Sie werden eingesetzt, um die Regeneration, Wiederherstellung oder den Ersatz von menschlichen Geweben zu erreichen. Das erste in der EU zugelassene Arzneimittel für neuartige Therapien (seit 2009) ist ein solches biotechnologisch bearbeitetes Gewebeprodukt: ChondroCelect™ der Firma TiGenix [13]. Es handelt sich um eine Suspension aus ex vivo kultivierten, autologen Chondrozyten (Knorpelzellen) zur Reparatur des Knorpels im Knie von Erwachsenen. Das Präparat wird für jeden einzelnen Patienten individuell hergestellt, indem dem Patienten gesundes Knorpelgewebe entnommen wird und die Chondrozyten expandiert (in vitro vermehrt) werden. Die Expansion stellt eine substanzielle Manipulation der Zellen dar, die in der Definition von biotechnologisch bearbeiteten Gewebeprodukten beschrieben ist. Sie dauert längere Zeit (bis zu vier Wochen, aber maximal drei Passagen laut europäischem öffentlichen Beurteilungsbericht der EMA) und geht mit einer gewissen Entdifferenzierung der Chondrozyten einher, sodass der Hersteller im Zulassungsverfahren unter anderem nachweisen musste, dass die Zellen sich nach der Applikation wieder zu vollwertigen Knorpelzellen redifferenzieren. Derartige Untersuchungen sind nur für neuartige Therapeutika mit substanzieller Manipulation, nicht aber für Gewebezubereitungen notwendig.
ATMP-Verordnung
Die ATMP-Verordnung schreibt für Arzneimittel für neuartige Therapien das zentralisierte Zulassungsverfahren vor [10]. Dies ist besonders wichtig für alle biotechnologisch bearbeiteten Gewebeprodukte, da diese bisher in einzelnen Mitgliedstaaten mit unterschiedlicher regulatorischer Basis in Verkehr waren. In Deutschland reichte bisher eine Herstellungserlaubnis der zuständigen Landesbehörde (in der Regel die Bezirksregierung) aus, wenn das Produkt nicht an andere abgegeben, sondern direkt angewandt wurde. Ähnlich war die Situation bei solchen somatischen Zelltherapeutika, die nicht im zentralisierten Verfahren zugelassen waren. (Die Verordnung (EG) Nr. 726/2004 schreibt die zentralisierte Zulassung von Arzneimitteln gegen Erkrankungen vor, bei denen jeder europäische Bürger die gleiche Chance der Behandlung haben sollte. Dazu gehören virale Infektionskrankheiten, Aids, Krebs, Diabetes, neurodegenerative Erkrankungen, Autoimmunerkrankungen und andere Immunstörungen).
Mit der ATMP-Verordnung wurde bei der Europäischen Arzneimittelbehörde (EMA) der Ausschuss Neuartige Therapien (Committee for Advanced Therapies, CAT) eingerichtet, der für die Bewertung der entsprechenden Zulassungsanträge zuständig ist. Zudem ist er an allen wissenschaftlichen Beratungen für ATMP beteiligt und betreut zwei für die ATMP neu geschaffenen regulatorischen Verfahren: die Klassifizierung von ATMP und die Zertifizierung von qualitätsbezogenen und nicht-klinischen Daten der ATMP-Entwicklung.
Da bereits einige biotechnologisch bearbeitete Gewebeprodukte und somatische Zelltherapeutika in einzelnen europäischen Mitgliedstaaten auf dem Markt waren, wurde jeweils eine Übergangsfrist festgelegt, in der diese Produkte eine zentralisierte Zulassung nachzuweisen haben. Diese endet bei den somatischen Zelltherapeutika am 30. 12. 2011, und bei den biotechnologisch bearbeiteten Gewebeprodukten am 30. 12. 2012.
Darüber hinaus wurden in der ATMP-Verordnung verschiedene Anreize und Vergünstigungen für die Hersteller von ATMP festgelegt, um die Entwicklung innovativer Präparate zu erleichtern. Am Paul-Ehrlich-Institut wurde ein Innovationsbüro etabliert, das Firmen in allen regulatorischen Fragen bei der Entwicklung von ATMP berät [14].
Zusammenfassung
Durch die verbesserten Lebensumstände in der westlichen Welt und die damit verbundene gestiegene Lebenserwartung ist ein Bedarf an der medizinischen Wiederherstellung von Körperfunktionen entstanden, der ständig steigt. Neben den Medizinprodukten der regenerativen Medizin und den Gewebespenden verspricht der sehr innovative Bereich der neuartigen Therapien, insbesondere der biotechnologisch bearbeiteten Gewebeprodukte (ATMP), einen großen Therapiefortschritt. Einige ATMP sind bereits in einzelnen Europäischen Mitgliedstaaten auf dem Markt, stehen allerdings nicht allen europäischen Bürgern zur Verfügung. Die Europäische Legislative hat daher mit der Gewebeverordnung und der ATMP-Verordnung einen harmonisierten Rechtsrahmen geschaffen, der künftig allen europäischen Bürgern gleichen Zugang zu vergleichbar guten Produkten ermöglicht.
Quellen [1] Richtlinie 2004/23/EG des Europäischen Parlaments und des Rates vom 31. 03. 2004. ABl. EU Nr. L 102/48 v. 07. 04. 2004. [2] Richtlinie 2006/86/EG der Kommission vom 24. 10. 2006. ABl. EU Nr. L 294/32 v. 25. 10. 2006. [3] Richtlinie 2006/86/EG der Kommission vom 08. 02. 2006. ABl. EU Nr. L 38/40 v. 09 .02. 2006. [4] http://ec.europa.eu/health/blood_tissues_organs/key_documents/index_en.htm#anchor6. [5] Gesetz über Qualität und Sicherheit von menschlichen Geweben und Zellen (Gewebegesetz) vom 20. 07. 2007. BGBl. I S. 1574. [6] Kloesel/Cyran, Arzneimittelrecht-Kommentar, Anm. 5 zu § 21a AMG. Stuttgart 2010. [7] Gesetz über die Spende, Entnahme und Übertragung von Organen und Geweben (Transplantationsgesetz – TPG) vom 04. 09. 2007 (BGBl. I S. 2206) i. d. geltenden Fassung. [8] Fuhrmann S, Klein B, Fleischfresser A (Hrsg). Arzneimittelrecht – Handbuch für die pharmazeutische Rechtspraxis. Stuttgart 2010, S. 1028 f. [9] Wie [8], S. 1009 f. [10] Verordnung (EG) Nr. 1394/2007 des Europäischen Parlaments und des Rates vom 13.11.2007 über Arzneimittel für neuartige Therapien […]. ABl. EU Nr. L 324/121 v. 10. 12. 2007. [11] Dingermann T, Zündorf I, Winckler T. Gentechnik, Biotechnik – Grundlagen und Wirkstoffe. 2. Aufl. Stuttgart 2011, S. 313 ff. [12] Caesar W. Therapeutischer Impfstoff Provenge zugelassen. DAZ.online v. 05. 05. 2010. [13] Weiß M. Symptomatische Knorpelschäden: Erste zellbasierte Arznei zugelassen. Dtsch Arztebl 2009;106(50):A‑2523. [14] www.pei.de/cln_092/nn_154420/DE/infos/pu/innovationsbuero/innovationsbuero-node.html?__nnn=true, E-Mail-Kontakt: innovation@pei.de.
Korrespondenzautorin
Prof. Dr. rer. nat. Christa Schröder
Fakultät Life Sciences, Studiengang Pharmatechnik
Hochschule Albstadt-Sigmaringen, Standort Sigmaringen



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.