













- DAZ.online
- DAZ / AZ
- DAZ 23/2010
- Von der Pathologie zur ...
Fortbildung
Von der Pathologie zur Pharmakotherapie
Gerd Geisslinger
Foto: DAZ/ck
Die Therapie darf aber kein Zufall sein, sondern sollte auf einer rationalen Pharmakotherapie basieren.
Der sogenannte physiologische Nozizeptorschmerz ist ein Warnsignal, um durch eine Reflexreaktion wie das Zurückziehen der Hand beim Berühren einer heißen Herdplatte einer Gewebsschädigung vorzubeugen. Er ist für das Überleben notwendig und sollte nicht unterdrückt werden.
Pathophysiologischer Nozizeptorschmerz
Der pathophysiologische Nozizeptorschmerz entsteht durch pathophysiologische Organveränderung bei einer Gewebsschädigung oder Entzündung. Er kann sich als Hyperalgesie, einer verstärkten Schmerzhaftigkeit auch sonst schon schmerzhafter Reize, oder als Allodynie, einer Schmerzempfindlichkeit normalerweise nicht schmerzhafter Reize, wie zum Beispiel der Berührungsschmerz bei einem Sonnenbrand, äußern. Die dritte Schmerzform, die neuropathischen Schmerzen, entstehen bei einer kontinuierlichen Schädigung peripherer Nerven nach Kompression, Durchtrennung, Entzündung oder einer metabolischen Störung wie Diabetes mellitus. Diese Schmerzen können lang anhaltend sein und haben ihren Warncharakter verloren.
Nach einer akuten Gewebsschädigung treten sofort Mediatoren aus, die über ihre Rezeptoren Nozizeptoren direkt erregen.
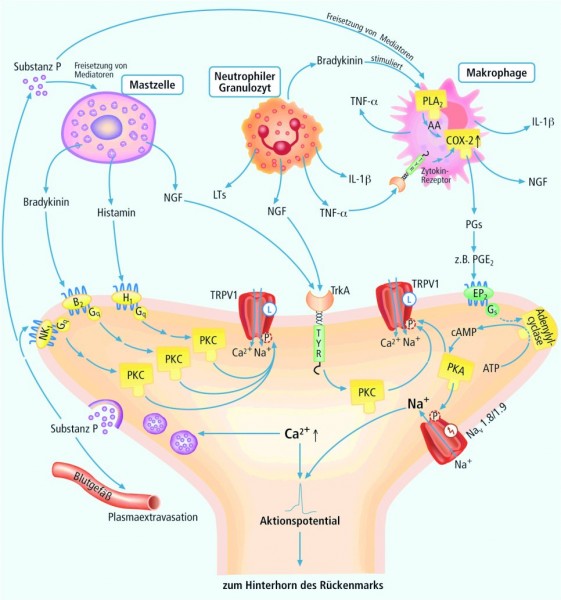
Bei einem akuten physiologischen Nozizeptorschmerz sind noch keine immunkompetenten Zellen beteiligt. Erst wenn sich nach einer Gewebsschädigung eine Entzündung entwickelt, führt dies zu einer Rekrutierung und Aktivierung von immunkompetenten Zellen, die nun ihrerseits Entzündungs- und Schmerzmediatoren synthetisieren und freisetzen. In einem komplexen Zusammenspiel werden so die Entzündungssymptome Ödem, Erythem, Schmerz und Hyperalgesie vermittelt. Mastzellen setzen den nerve growth factor (NGF) und Histamin frei. Bradykinin, das im Plasma aus Kininogen gebildet, gelangt ins entzündete Gewebe, bindet an B1- und B2-Rezeptoren, diese aktivieren die Proteinkinase C. In Folgeschritten wird auch die De-novo-Synthese von Prostaglandinen stimuliert. Neutrophile Granulozyten und Makrophagen produzieren Zytokine (TNF α und IL-1β), diese binden an ihre Rezeptoren und bewirken eine Aktivierung der Transkription von inflammatorischen Genen z. B. Cyclooxygenase 2 (COX-2). Die Cyclooxygenase ist aus einem pharmakotherapeutischen Blickwinkel einer der wichtigsten Beteiligten am Schmerzgeschehen. COX-2 katalysiert die Umwandlung von Arachidonsäure in Prostaglandine. Folge ist eine überschiessende Produktion von Prostaglandinen.
Ist weniger manchmal mehr?
Prostaglandine sensibilisieren in der Peripherie direkt Nozizeptoren über eine Phosphorylierung von TRPV1- und Natriumkanälen. Auf Rückenmarksebene hemmt PGE2 inhibitorische Interneurone und führt somit zu einer verstärkten Schmerzweiteleitung. Der Hemmung der Prostaglandinsynthese kommt daher bei der Therapie entzündlicher Schmerzen eine große Rolle zu. Sie ist über zwei Mechanismen möglich: Steroidale Antiphlogistika hemmen die induzierte Prostaglandinsynthese, indem sie die entzündungsbedingte Induktion der COX-2 hemmen, die basale Prostaglandinsynthese wird nicht gehemmt. Nicht-steroidale Antiphlogistika (NSAID) hemmen die Cyclooxygenasen in ihrer Funktion und damit die Prostaglandinsynthese. Da Prostaglandine zahlreiche physiologische Funktionen wahrnehmen, gehen mit einer Hemmung und der gewünschten antientzündlichen Wirkung auch unerwünschte Wirkungen einher. Vor allem bei den nicht-selektiven NSAID gehören dazu u.a. gastrointestinale Störungen und Hautreaktionen ebenso wie Nierenfunktionsstörungen. Alle nicht-steroidalen Antiphlogistika, nicht-selektive NSAID und selektive COX-2-Hemmer, erhöhen das Risiko für kardiovaskuläre Ereignisse. Schätzungsweise treten drei bis vier Fälle/1000 Patienten/Jahr auf. Als Ausnahmen nannte Geisslinger niedrig dosierte Acetylsalicylsäure und hoch dosiertes Naproxen (1000 mg/d), die beide zu einer messbaren Thrombozytenaggregationshemmung führen. Bei selektiven COX-2-Hemmer wie Celecoxib, Etoricoxib oder Parecoxib treten ca. 50% weniger gastrointestinale Komplikationen auf. Neben der klaren proinflammatorischen Rolle der Prostaglandine wird diskutiert, ob sie auch in der späten Phase einer Entzündung für die Resolution, die Auflösung der Entzündung, bedeutsam sind. Geisslinger stellte die Frage in den Raum, ob NSAIDs die physiologische Auflösung einer Entzündung teilweise sogar verhindern können? Er beobachtete, dass NSAIDs bei chronischen Schmerzen nicht so gut wirken und oft den Patienten keinen Vorteil bringen. Laborversuche deuten eine Erklärung an: In Zellkulturen an Mikrogliazellen, mit denen eine chronische Inflammation modellhaft untersucht wird, konnte gezeigt werden, dass PGE2 über eine negative Feedback-Reaktion die eigene Synthese hemmen kann. Langfristig scheint PGE2 eine duale Wirkung zu haben: akut inflammatorisch aktiv, chronisch antiinflammatorisch aktiv. Wird bei einer chronischen Entzündung die Prostaglandinsynthese mittels NSAID unterdrückt, so könnte auch die antiinflammatorische PGE2 -Komponente inhibiert werden. Dies würde erklären, warum bei längerer Einnahme die Wirkung der NSAID nachzulassen scheint. Geisslinger riet, bei Bedarf ein bis zwei Wochen NSAIDs zu geben und dann zu pausieren. ck
Bei entzündlichen Schmerzen
werden u. a. neutrophile Granulozyten, Makrophagen und Mastzellen aktiviert, die bestimmte Schmerzmediatoren freisetzen und so entscheidend zum entzündlichen Geschehen beitragen.
(NGF: nerve growth factor; TNF a Tumornekrosefaktor alpha, IL-1 Interleukin 1, LTs Leukotriene, PLA2
Phospholipase A2,
AA Arachidonsäure, COX-2 Cyclooxygenase 2, PGs Prostaglandine, EP2
Prostaglandin-E-Rezeptor vom Typ 2, cAMP cyclisches Adenosinmonophosphat, PKA Proteinkinase A, TrkA Tyrosinkinase-Rezeptor, PKC Proteinkinase C, NK1
Neurokinin-1-Rezeptor)
Quelle: Mutschler, E.: Arzneimittelwirkungen. Lehrbuch der Pharmakologie und Toxikologie.
9. Auflage,
Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart (2008).
9. Auflage,
Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart (2008).



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.