













- DAZ.online
- DAZ / AZ
- DAZ 20/2010
- Vorteile gegenüber ...
Schmerztherapie
Vorteile gegenüber oraler Schmerztherapie?
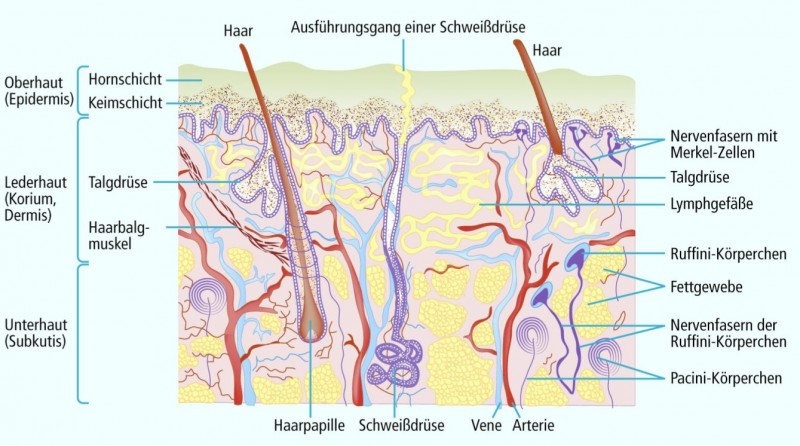
Abb. 1: Schematische Darstellung der menschlichen äusseren Haut
[nach Thews/Mutschler/Vaupel] Transdermal applizierte Wirkstoffe
müssen eine Reihe hydrophiler und lipophiler Schichten überwinden, bevor sie Wirkorte (feine Nervenendigungen in Haut und Synovia) erreichen können. Das dichte, subkutane Kapillarnetz macht die direkte Diffusion sinnvoller Mengen von Cyclooxygenasehemmern direkt in das entzündete Gelenkgewebe unwahrscheinlich. Plausibler ist die Aufnahme in das Kapillarbett und der Transfer in das Gelenk über den Blutweg.
Unsere Haut dient primär dazu, uns von der Umwelt abzuschirmen und das Eindringen von Fremdmolekülen in den Körper zu verhindern. So muss jede Substanz, die transdermal appliziert wird und unterhalb der oberen Hautschichten (Dermis) ihre Wirkung entfalten soll, die wichtige lipophile Barriere (Stratum corneum) überwinden [8]. Der alternative hydrophile Weg (z. B. über die Schweißdrüsen) ist quantitativ nur begrenzt nutzbar (vgl. Abb. 1). Wirkstoffe, die das Stratum corneum überwunden haben, treffen im Unterhautgewebe auf zahlreiche Kapillaren, Lymphgefäße, Bindegewebs-, Fett- und Nervenzellen. Hydrophilere Substanzen werden, so sie denn bis in diesen Bereich vordringen, in das Kapillarnetz diffundieren und abtransportiert werden – oft bevor sie die Zielstrukturen (z. B. Nozizeptoren in der Gelenksynovialis; [14]) erreicht haben. Sehr lipophile Substanzen können in die Lipide z. B. der Fettzellen eingelagert und nur langsam wieder freigesetzt werden. In jedem Fall ist die Diffusion in tiefere Strukturen, z. B. Gelenke, nicht ohne Weiteres vorstellbar, da lipophile und hydrophile Pharmaka das dichte Kapillarnetz und das Fettgewebe quasi umschiffen müssten, um direkt, d. h. unter Umgehung des Blutkreislaufs, in tiefere Strukturen vorzudringen. Unabhängig vom Diffusionsweg kann davon ausgegangen werden, dass die transdermale Absorption ein zögerlicher Prozess ist, der Spitzenkonzentrationen im Plasma genauso wie z. B. in der Gelenkflüssigkeit nur nach längerer Latenzzeit (Stunden – Tage) erlaubt [6, 15, 21, 25, 26]. Eine akute Schmerzlinderung ist daher weder durch NSAR noch durch Opioide zu erwarten.
Anders ist die Situation bei der Anwendung von Lokalanästhetika. Diese können hydrophile und lipophile Eigenschaften in sich vereinigen und ihr Zielsubstrat, nämlich feine, sensible Nervenendigungen, die sich direkt unterhalb der Epidermis befinden (Abb. 1), schnell erreichen, vor allem bei Injektion in die Subcutis. Sie werden hier nicht weiter diskutiert.
Schließlich ergibt sich aus den anatomisch-physiologischen Überlegungen, dass eine hohe transdermale Bioverfügbarkeit eines Wirkstoffes zur systemischen Wirkung führt, wenn genügend Wirkstoff auf die Haut aufgetragen wird. Dies impliziert, dass die bekannten, unerwünschten Arzneimittelwirkungen grundsätzlich auch bei transdermaler Applikation auftreten können – vorausgesetzt, dass adäquate Mengen absorbiert und systemisch wirksame Konzentrationen erreicht werden.
NSAR: topischer Effekt denkbar
NSAR antagonisieren durch Hemmung der Bildung von Prostaglandinen im (entzündeten) schmerzhaften Gewebe (z. B. Synovialis) und im ZNS die schmerzhafte Hyperalgesie [14]. Grundsätzlich ist daher ein topischer, analgetischer Effekt denkbar [5]. Aus den anatomisch-physiologischen Überlegungen ergibt sich, dass nur sehr lipophile Cyclooxygenasehemmer zur erfolgreichen transdermalen Applikation zur Verfügung stehen.
Tab. 1: Häufig systemische, phototoxische und photoallergische Reaktionen auslösende Wirkstoffe [nach 2] |
Nicht-steroidale Antiphlogistika |
Benoxaprofen |
Piroxicam |
Carprofen |
Tiaprofensäure |
Naproxen |
Ketoprofen |
Cave Phototoxizität
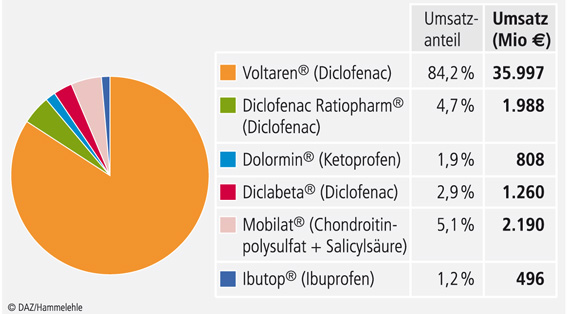
Sie müssen darüber hinaus lichtstabil sein und dürfen nicht zu phototoxischen Reaktionen in der Haut führen [2, 7, 16]. Substanzen mit hohem phototoxischem Potenzial: (vgl. Tab. 1), wie z. B. Benoxaprofen (nicht mehr zugelassen), Carprofen (nur noch in der veterinärmedizinischen Anwendung) und Ketoprofen, müssen daher als wenig geeignet gelten. Andere Wirkstoffe erscheinen zu wenig potent, um transdermal Wirkungen entfalten zu können. Dazu gehören die Salicylate und das Paracetamol. Ob bestimmte Salicylate durch lokale Irritation im Sinne der "counterirritation" analgetische Wirkungen auslösen können, ist fraglich [1, 19]. Ibuprofen ist aufgrund seiner geringen analgetischen Potenz kein Mittel der ersten Wahl [18, 20, 26]. Gegen Diclofenac, Flurbiprofen und einige Andere bestehen nur geringe Bedenken, obwohl auch für diese Wirkstoffe phototoxische Ereignisse beschrieben sind [20]. Auch Indomethazin und Flufenaminsäure könnten geeignet sein. In der Tat gibt es transdermale Präparationen, die die Lipophilie z. B. der Flufenaminsäure durch Veresterung der Säuregruppe erhöhen (z. B. Etofenamat) und damit zu einer relativ hohen transdermalen Bioverfügbarkeit führen. Allerdings fehlen für diese Präparate wissenschaftlich belastbare, placebokontrollierte Doppelblindstudien zur Wirksamkeit [27]. Dieser Analyse geeigneter Wirkstoffe für die transdermale Applikation entsprechen die Umsätze in Deutschland. Diclofenac lässt andere Cyclooxygenasehemmer mit über 90% Marktanteil weit hinter sich (Abb. 2).
Diclofenac-Wirkung durch COX-Hemmung?
Von den genannten Wirkstoffen wurde nur für Diclofenac in mehreren Doppelblindstudien eine gewisse Wirksamkeit bei muskulo-skelettalen Schmerzen demonstriert [vgl. 3, 9, 18, 19, 26]. Wie diese Wirksamkeit zustande kommt, ist umstritten. Es gibt keine zweifelsfreien Belege dafür, dass Cyclooxygenasehemmung durch direkte Diffusion von der Haut hinein in z. B. arthrotisch veränderte Gelenke, wie Knie- oder Hüftgelenk, möglich ist.
Im Gegenteil: Da NSAR-Topika typischerweise am entzündeten (traumatisierten) Gelenk appliziert werden und dort höhere Konzentrationen erreichen als im nicht entzündeten Kontrollgelenk, wurde oft auf eine direkte Diffusion in das Gelenk geschlossen.
Diffusion von der Haut ins Gelenk?
Die Untersuchungen von Radermacher et al. belegen allerdings, dass die hohen Konzentrationen im entzündeten Gelenk hämatogen sind [2]. Die Autoren applizierten Diclofenac-Gel bei Patienten mit zwei etwa gleich stark entzündeten Kniegelenken – entweder auf das rechte oder linke Knie. Danach wurden beide Gelenke punktiert, und es wurde die Diclofenac-Konzentration im Plasma und in der Gelenkflüssigkeit bestimmt. Die freien Konzentrationen waren in beiden Gelenken etwa gleich, aber etwas geringer als im Plasma – unabhängig davon, an welchem Knie das Gel appliziert wurde. Es fand also keine wesentliche Diffusion von der Haut ins Kniegelenk statt [21]. Andere Untersuchungen zeigen, dass die Gesamtkonzentration von Diclofenac auch bei oraler Gabe im entzündeten Gewebe höher ist als im gesunden Gewebe [5]. Die subkutane Muskulatur, die subkutanen Sehnen sowie kleine, oberflächennahe Gelenke können allerdings durch direkte Diffusion erreicht werden [22].
Gleiche Wirkung bei weniger Nebenwirkungen?
Von größerer Bedeutung scheint es, die immer wieder gemachte Behauptung zu analysieren, die transdermale Applikation sei bei "gleicher Wirksamkeit" mit deutlich weniger unerwünschten Arzneimittelwirkungen verquickt. Diese Vermutung [vgl. z. B. 1, 9, 17] ist nicht überzeugend belegt. Bei der oralen Gabe auch niedriger Dosen ist Diclofenac zu ca. 50% bioverfügbar [11]. Bei der topischen Applikation von z. B. 100 mg Diclofenac sind nur ca. 5 bis 8% im Vergleich zur oralen Gabe bioverfügbar [12, 15]. Dass dieses ausreicht, eine temporäre Reduktion der Prostaglandin-E2-Synthese ex vivo zu vermitteln, ergibt sich aus der Abbildung 3. Eine vergleichbare systemische Prostaglandinsynthesehemmung ist auch mit etwa 5 bis 10 mg Diclofenac erzielbar – die dann allerdings ebenfalls kaum die Diclofenac-typischen, unerwünschten Arzneimittelwirkungen, wie gastrointestinale Störungen, Wasser- und Elektrolytretention, Hemmung der Blutgerinnung, auslösen würde. Schließlich fehlt bei solchen Dosen eine nachhaltige Hemmung der Cyclooxygenasen [12, 15]. Entsprechende Untersuchungen (z. B. der Vergleich von 100 mg/d Diclofenac transdermal vs. 5 – 10 mg/d p.o.) gibt es nicht.
Diclofenac oral und topisch: ein Vergleich
Eine kürzlich erschienene, methodisch sehr durchdachte Studie [23] vergleicht 100 mg Diclofenac p.o. mit etwa 72 mg Diclofenac transkutan. Dazu wurden viermal täglich 1,2 ml Diclofenac-haltige DMSO-Lösung auf das schmerzende Gelenk aufgetragen bzw. 100 mg oral eingenommen (Anmerkung: in Deutschland erhältliche topische Diclofenac-Zubereitungen enthalten kein DMSO). Beide Diclofenac-Gaben waren im Vergleich zu Placebo (gering, aber signifikant) wirksam. Unterschiede zwischen oraler und transdermaler Zufuhr konnten statistisch nicht gesichert werden. Allerdings bedeutet das Fehlen eines signifikanten Wirksamkeitsunterschieds zwischen topischer und oraler Diclofenac-Gabe nicht die therapeutische Gleichwertigkeit (non-inferiority). Aufgrund der geringen systemisch wirksamen Dosis (ca. 5 – 10 mg) in dieser Studie ist die geringe Inzidenz von gastrointestinalen Nebenwirkungen nicht verwunderlich. Dafür traten zahlreiche topische Probleme nach lokaler Applikation auf, die evtl. auf das verwendete DMSO-Lösungsmittel zurückzuführen sind [23].
Transdermale Applikation: zusätzliche Risiken
Die transdermale Applikation ist mit zusätzlichen Risiken verbunden. Grundsätzlich kann es zu allergischen Reaktionen gegen den Wirkstoff oder gegen in der Präparation vorhandene Hilfsstoffe kommen. Besonders bedenklich scheinen Pflaster (Patches) zu sein. Erfahrungen aus der transdermalen (Pflaster-) Applikation von Opioiden weisen darauf hin, dass topische Infektionen und toxische Reaktionen bei fast 10% der Patienten auftreten [6]. Ungeklärt bleibt die Frage, ob eine transdermale Applikation von NSAR zur Sensibilisierung führen kann, die später bei oraler oder parenteraler Applikation des Wirkstoffes unterschiedliche immun-allergische/hämatologische Probleme auslösen [4].
FDA-Warnung vor Leberschäden
So warnt die FDA auch bei transdermaler Applikation von Diclofenac vor Leberschäden. Der Arzneimittelkommission der Deutschen Ärzteschaft liegen einige wenige Berichte über Transaminasenerhöhungen im Zusammenhang mit der topischen Gabe von Diclofenac vor. Der Hersteller teilt auf Anfrage mit, dass er kein reales Risiko identifizieren kann [29].
Opioide: lipophile Arzneistoffe scheinen geeignet
Für die Auswahl der Wirkstoffe aus der Gruppe der Opioide für die transdermale Applikation gilt grundsätzlich Ähnliches wie bei den Cyclooxygenasehemmern. Hoch potente, relativ lipophile Wirkstoffe scheinen zur transdermalen Applikation geeignet. Von therapeutischer Bedeutung sind in Deutschland intensiv verwendete Pflasterapplikationen von Fentanyl (z. B. Durogesic®) und Buprenorphin (z. B. Transtec®). Hier wird ein Nachteil beider Wirkstoffe kompensiert, bei oraler Applikation im Magen-Darm-Trakt und während der primären Leberpassage praktisch vollständig inaktiviert zu werden. Fentanyl und Buprenorphin sind daher zurzeit nur bei parenteraler, rektaler oder sublingualer Applikation zur Dauertherapie verwendbar.
Opioide: keine topische Analgesie
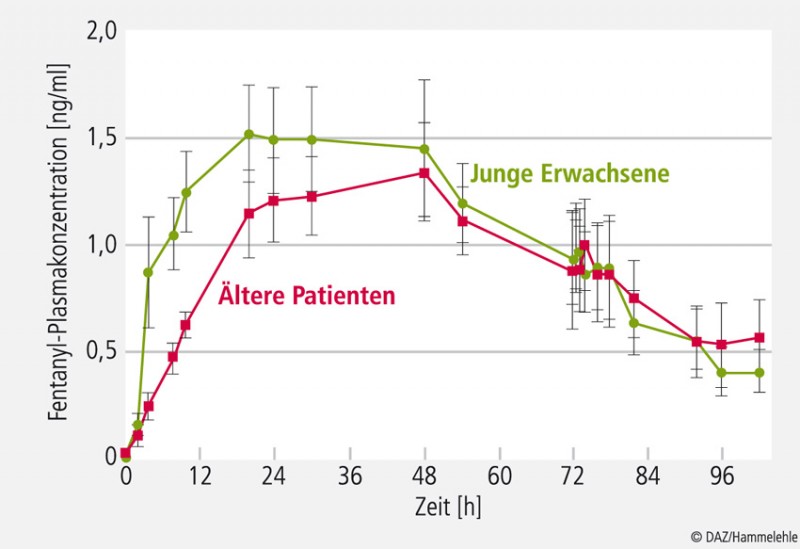
Für die Gruppe der Opioide wird keine topische, analgetische Wirkung postuliert. Sie wirken nicht im geschädigten (schmerzhaften) Organsystem (z. B. Gelenk), sondern finden ihren Hauptwirkort im ZNS [14]. Es besteht kein Zweifel, dass eine systemische Wirksamkeit von Fentanyl und Buprenorphin bei transdermaler Applikation möglich ist. Dementsprechend gelten für die transdermale Applikation dieser Wirkstoffe dieselben Risiken wie für andere parenterale Applikationsformen. Zusätzliche Probleme resultieren aus Schäden am Applikationsort und aus der komplexen Pharmakokinetik [22, 25] bei dieser Anwendung (vgl. Abb. 4).
Transdermale Opioide: zusätzliche Risiken
Die Pharmakokinetik erklärt, warum z. B. Fentanyl transdermal nicht sofort nach Applikation wirkt. Es wird zunächst in den subkutanen Fettspeichern und anderen Hautstrukturen "zwischengelagert". Erst wenn (später) genügend Wirkstoff in diesen Depots vorhanden ist, wird er in das Blut abgegeben, und es beginnt die analgetische Wirkung, die ihr Maximum erst ca. 12 bis 36 Stunden nach Pflasterapplikation erreicht (Abb. 4; [22, 25]). Demzufolge werden die Wirkungen und die damit einhergehenden Nebenwirkungen, wie z. B. Atmungshemmung, ebenfalls erst spät auftreten. Sie können auch nach Pflasterentfernung noch mehrere Tage anhalten, bis die kutanen Speicher wieder entleert sind.
Verschärft wird diese problematische Situation dadurch, dass je nach Hautstruktur, -temperatur und -durchblutung unterschiedlich schnell und unterschiedlich viel Wirkstoff in den Gesamtorganismus aufgenommen wird. Zahlreiche akut lebensbedrohliche, unerwünschte Arzneimittelwirkungen resultieren aus der schwierigen richtigen Dosierung und Anwendung von Wirkstoffen, die nicht akut schmerzlindernd wirken, sondern nur langsam zur Schmerzhemmung führen, weil sie in äußerst komplexer Weise aufgenommen, gespeichert, verteilt und eliminiert werden.
Tödliche Überdosierung bei Fehlanwendung
Die hohe Wirksamkeit der transdermal verwendeten Opioide, ihre komplexe Verstoffwechselung und die bei transdermaler Applikation variable und unübersichtliche Pharmakokinetik (Anflutung, Spitzenkonzentration und Elimination) scheinen die Ursache für die zahlreichen Meldungen über unerwünschte Arzneimittelwirkungen in Deutschland und anderen Ländern zu sein. Die Opioid-typischen Probleme (Übelkeit, Benommenheit, Somnolenz, Erbrechen) stehen quantitativ im Vordergrund [6]. Berichtet werden aber auch die schwerwiegenderen Atmungsstörungen und Anomalien bis hin zur Schnappatmung. Schließlich kommen Todesfälle zur Meldung, bei denen offensichtlich eine massive Überdosierung stattgefunden hat – entweder durch irrtümliche Anwendung von Fentanyl-Pflastern mit hoher Freisetzungsrate bei zierlichen Patienten bei fehlender Gewöhnung an Opiode oder durch eindeutige Anwendungsfehler, weil falsche Pflasterstärken oder Mehrfachpflasterungen zur gleichen Zeit zum Einsatz kommen. Eine Reihe von Fallschilderungen, die diese Problematik dokumentieren, finden sich z. B. in [24].
Schließlich muss auch im Zusammenhang mit den Opioiden auf topische Probleme hingewiesen werden. Hautirritationen, mikrobieller Befall im Bereich der Pflasterstellen, allergische Reaktionen gegen Wirk- oder Klebstoff sind nicht die Ausnahme, sondern häufige Begleiterscheinungen dieser Therapie.
Schlussfolgerung
Wie eingangs ausgeführt, ist die zurzeit modische Applikation von analgetischen Wirkstoffen transdermal nur selten eine überzeugende therapeutische Option. Die für die Cyclooxygenasehemmer postulierte, direkte Vermittlung von analgetischen Wirkungen im geschädigten Organsystem, z. B. in arthrotischen Gelenken, erscheint nur für wenige Wirkstoffe und wenige Arthrosen (Fingergelenke) plausibel und belegt.
Die immer wieder behauptete geringere Inzidenz von Wirkstoff-typischen Nebenwirkungen (Magen-Darm-Trakt) lässt sich durch die geringen systemisch verfügbaren Mengen der Wirkstoffe erklären. Eine sehr gute Verträglichkeit von z. B. Diclofenac könnte demnach vermutlich auch durch sehr niedrige orale Dosen erzielt werden. Damit würden zwar die verzögerte analgetische Wirkung, der späte Wirkungseintritt und die topischen Nebenwirkungen vermieden, aber die psychologische Zuwendung und der hohe Placebowert der transdermalen Anwendung würden vielen Patienten sicher abgehen.
Ungeklärt bleibt, in welchem Umfang eine transdermale Applikation zur immunologischen Sensibilisierung führt, die später akute hepatische, kutane oder hämatopoetische Probleme provozieren kann.
Anders, aber ähnlich unbefriedigend ist die Situation bei der transdermalen Applikation von Opioiden. Die oft erwartete, sofortige Schmerzlinderung muss – systembedingt – fehlen. Die Dosierung ist daher individuell sehr schwierig: Der Wirkungseintritt erfolgt spät, und wann das Wirkungsmaximum erreicht wird und wie ausgeprägt es ist, bleibt im Einzelfall nicht voraussehbar. Die lange Verweildauer der Wirkstoffe in Fettdepots der Haut macht die Anwendung anderer Analgetika problematisch, da es zu Potenzierungsvorgängen (z. B. der Atmungshemmung) kommen kann. Schließlich haben die für die kutane Anwendung notwendigen, speziellen galenischen Präparationen, wie Pflaster etc., ihre eigenen Probleme. Sie führen zu topischen und allergischen Reaktionen auch gegen Hilfsstoffe. Sie begünstigen das Wachstum von Hautparasiten, wie Bakterien und Pilzen, unter den Pflastern. Die zahlreichen Meldungen von schweren Zwischenfällen bei der Anwendung transdermaler Fentanyl- und Buprenorphin-Pflaster belegen, wie problematisch dieser Ansatz ist.
Unabhängigkeitserklärung
Der Autor hat Vortragshonorare, Forschungsunterstützungen und Beratungshonorare von den Firmen Aventis, Bayer, GSK, Merck, Sharp & Dohme, Novartis und Pfizer erhalten.
Autor
Prof. Dr. med. Dr. h.c. Kay Brune
Doerenkamp Professor
FAU Erlangen-Nürnberg
Fahrstr.
91054 Erlangen
Literatur [1] Altman R, Barkin RL. Topical therapy for osteoarthritis: clinical and pharmacologic perspectives. Postgrad Med. 2009 Mar;121(2):139 – 47. [2] AWMF online. Leitlinien der Deutschen Dermatologischen Gesellschaft (DDG): Phototoxische und photoallergische Reaktionen. AWMF Leitlinien-Register Nr. 013/035. [3] Barthel HR, Haselwood D, Longley S 3rd, Gold MS, Altman RD. Randomized controlled trial of diclofenac sodium gel in knee osteoarthritis. Semin Arthritis Rheum. 2009 Dec;39(3):203 –12. [4] Boelsterli UA. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol. 2003 Nov 1;192(3):307 – 22. [5] Brune K, Furst DE. Combining enzyme specificity and tissue selectivity of cyclooxygenase inhibitors: towards better tolerability? Rheumatology (Oxford). 2007 Jun;46(6):911 – 9. [6] Durogesic(R) SMAT. Fachinformation. Oktober 2008. [7] Encinas S, Bosca F, Miranda MA. Phototoxicity associated with diclofenac: a photophysical, photochemical, and photobiological study on the drug and its photoproducts. Chem Res Toxicol. 1998 Aug;11(8):946 – 52. [8] Flynn GL. Mechanism of Percutaneous Absorption from Physicochemical Evidence. In: Dermatology. Volume 6: Percutaneous Absorption: Mechanisms – Methodologoy – Drug Delivery. Ed. Bronaugh RL, Maibach HI. Marcel Dekker, Inc. New York 1985, 17 – 42. [9] Gøtzsche PC. Non-steroidal anti-inflammatory drugs. BMJ. 2000 Apr 15;320(7241):1058 – 61. [10] Hinz B, Brune K. Can drug removals involving cyclooxygenase-2 inhibitors be avoided? A plea for human pharmacology. Trends Pharmacol Sci. 2008 Aug;29(8):391 – 7. [11] Hinz B, Chevts J, Renner B, Wuttke H, Rau T, Schmidt A, Szelenyi I, Brune K, Werner U. Bioavailability of diclofenac potassium at low doses. Br J Clin Pharmacol. 2005 Jan;59(1):80 – 4. [12] Hui X, Hewitt PG, Poblete N, Maibach HI, Shainhouse JZ, Wester RC. In vivo bioavailability and metabolism of topical diclofenac lotion in human volunteers. Pharm Res. 1998 Oct;15(10):1589 – 95. [13] Huntjens DR, Danhof M, Della Pasqua OE. Pharmacokinetic-pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology (Oxford). 2005 Jul;44(7):846 – 59. [14] Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001 Sep 13;413(6852):203 –10. [15] Kienzler JL, Gold M, Nollevaux F. Systemic bioavailability of topical diclofenac sodium gel 1% versus oral diclofenac sodium in healthy volunteers. J Clin Pharmacol. 2010 Jan;50(1):50 – 61. [16] Lichtsensibilisierung durch Arzneimittel. Arzneimitteltelegramm 1995:7:70 – 2. [17] Makris UE, Kohler MJ, Fraenkel L. Adverse Effects of Topical Nonsteroidal Antiinflammatory Drugs in Older Adults with Osteoarthritis: A Systematic Literature Review. J Rheumatol. 2010 Apr 1. [18] Mason L, Moore RA, Edwards JE, Derry S, McQuay HJ. Topical NSAIDs for chronic musculoskeletal pain: systematic review and meta-analysis. BMC Musculoskelet Disord. 2004 Aug 19;5:28. [19] Matthews P, Derry S, Moore RA, McQuay HJ. Topical rubefacients for acute and chronic pain in adults. Cochrane Database Syst Rev. 2009 Jul 8;(3):CD007403. [20] Moore DE. Mechanisms of photosensitization by phototoxic drugs. Mutat Res. 1998 Nov 9;422(1): 165 – 73. [21] Radermacher J, Jentsch D, Scholl MA, Lustinetz T, Frölich JC. Diclofenac concentrations in synovial fluid and plasma after cutaneous application in inflammatory and degenerative joint disease. Br J Clin Pharmacol. 1991 May;31(5):537 – 41. [22] Riess W, Schmid K, Botta L, Kobayashi K, Moppert J, Schneider W, Sioufi A, Strusberg A, Tomasi M. [The percutaneous absorption of diclofenac] Arzneimittelforschung. 1986 Jul;36(7):1092 – 6. [23] Simon LS, Grierson LM, Naseer Z, Bookman AA, Zev Shainhouse J. Efficacy and safety of topical diclofenac containing dimethyl sulfoxide (DMSO) compared with those of topical placebo, DMSO vehicle and oral diclofenac for knee osteoarthritis. Pain. 2009 Jun;143(3):238 – 45. [24] Stammschulte T, Brune K. [Drug safety problems in association with the use of opioid containing patches for the management of pain] Dtsch Med Wochenschr. 2010 Apr;135(17):870 – 3. [25] Thompson JP, Bower S, Liddle AM, Rowbotham DJ. Perioperative pharmacokinetics of transdermal fentanyl in elderly and young adult patients. Br J Anaesth. 1998 Aug;81(2): 152 – 4. [26] Underwood M, Ashby D, Carnes D, Castelnuovo E, Cross P, Harding G, Hennessy E, Letley L, Martin J, Mt-Isa S, Parsons S, Spencer A, Vickers M, Whyte K. Topical or oral ibuprofen for chronic knee pain in older people. The TOIB study. Health Technol Assess. 2008 May;12(22):iii-iv, ix-155. [27] Vanderstraeten G, Schuermans P. Study on the effect of etofenamate 10% cream in comparison with an oral NSAID in strains and sprains due to sports injuries. Acta Belg Med Phys. 1990 Jul-Sep;13(3): 139 – 41. [28] Watson MC, Brookes ST, Kirwan JR, Faulkner A. Non-aspirin, non-steroidal anti-inflammatory drugs for osteoarthritis of the knee. Cochrane Database Syst Rev. 2000;(2):CD000142. [29] Antwort der Firma Novartis auf Anfrage nach Leberschäden im Zusammenhang mit der topischen Applikation: "Novartis hat keine spezifischen, sicherheitsrelevanten Trends im Sicherheitsprofil von Voltaren Schmerzgel/Voltaren Emulgel, wie etwa eine Erhöhung der Leberwerte, identifiziert. Seit der ersten Zulassung im Oktober 1985 haben mehr als 430 Millionen Patienten von Novartis Pharma und Novartis Consumer Health vermarktete topische Diclofenac Präparate weltweit angewendet (bis Dezember 2009). Die niedrige systemische Verfügbarkeit, Leberfunktionstests in Placebo-kontrollierten klinische Studien mit Behandlungszeiten bis zu 12 Wochen (mit Diclofenac-Natrium Gel), und die vorliegenden Anwendungserfahrungen bestätigen, dass Voltaren Schmerzgel/Voltaren Emulgel bei bestimmungsgemäßer Anwendung sicher sind."



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.