










- DAZ.online
- DAZ / AZ
- DAZ 4/2008
- Hoffen auf eine Alzheimer...
Alzheimer-Demenz
Hoffen auf eine Alzheimer-Impfung
Die Alzheimer-Erkrankung ist die häufigste neurodegenerative Erkrankung mit einer Prävalenz von 812/100.000 Personen und führt langsam, aber unaufhaltsam zum Verlust der Gedächtnisleistung und weiterer kognitiver Funktionen. Derzeit stehen nur symptomatische Therapieoptionen zur Verfügung. Hierzu gehören die Gruppe der Cholinesterase-Inhibitoren sowie der NMDA-Antagonist Memantine. In Deutschland sind als Cholinesterase-Inhibitoren Donepezil, Galantamin und Rivastigmin für die Behandlung der leichten und mittelschweren Demenz zugelassen [1, 2]. Ihr primärer Wirkmechanismus besteht darin, die Verfügbarkeit von Acetylcholin durch Hemmung des abbauenden Enzym Cholinesterase an den cholinergen Synapsen zu erhöhen. Für die Behandlung mittelschwerer bis schwerer Alzheimer-Demenz ist in Deutschland der N-Methyl-D-Aspartat(NMDA)-Rezeptorantagonist Memantine zugelassen, der seine Wirkung als nicht kompetitiver niederaffiner NMDA-Rezeptor-Antagonist entfaltet.
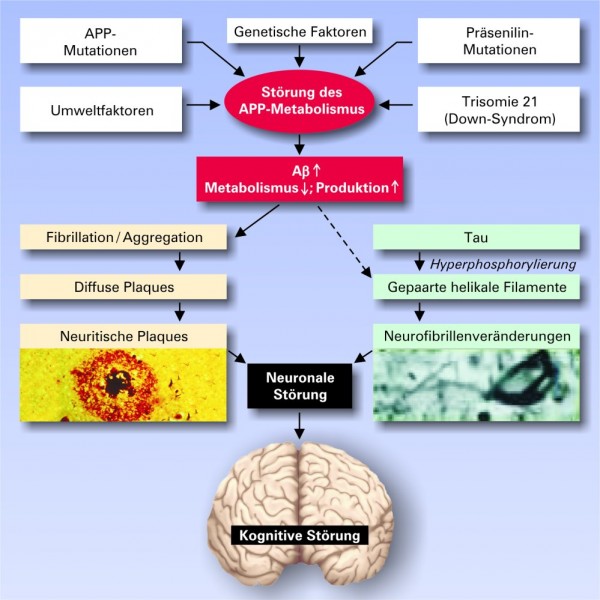
Da die Wirkung der derzeit zur Verfügung stehenden Behandlungsoptionen zeitlich begrenzt ist, werden derzeit eine Vielzahl von Therapieansätzen in präklinischen und klinischen Studien entwickelt (Tab. 1) [3]. Die derzeit vielversprechendsten Ansätze zielen auf die Beeinflussung der Amyloid-Kaskade, jedoch finden sich auch andere Ansätze, die in Tabelle 1 zusammengestellt sind. Um diese Therapieansätze besser verstehen zu können, soll in Kürze die Entstehungshypothese der Alzheimer-Krankheit beschrieben werden (Abb. 1).
Tab. 1: Experimentelle Therapieoptionen, die derzeit für die Behandlung der Alzheimer-Krankheit entwickelt oder getestet werden [3] | |
Wirkmechanismus |
Substanzen |
Antiamyloid-Therapieansätze |
Inhibitoren/Modulatoren der β- und γ-Sekretase
Aktivatoren der α-Sekretase
Hemmung der Aβ-Plaque-Bildung
Hemmung der Oligomerisierung/Fibrillierung
Statine
PPAR-γ Agonisten
Muskarinerge M1-Agonisten
|
Neuroprotektive Therapieansätze |
Nervenwachstumsfaktoren
Antioxidanzien
Astrozytenmodulatoren
Homocystein-reduzierende Faktoren
Anti-inflammatorische Substanzen
NMDA-Rezeptor-Antagonisten
Ampakine
Tau-bezogene Ansätze
Caspase-Inhibitoren
MAO-Inhibitoren
Cholinesterase-Inhibitoren
Nicotin-Acetylcholin-Rezeptor-Agonisten
|
Neurorestaurative Therapieansätze |
Neurotrophin
Zelltransplantation
|
Stammzellforschung |
|
Abb. 1:
Entstehungshypothese der Alzheimer-Erkrankung.
PS: Präsenilin; APP: Amyloid Präkursor Protein
Die Entstehungshypothesender Alzheimer-Krankheit
Histopathologisch ist die Alzheimer-Krankheit durch folgende Veränderungen charakterisiert:
- Verlust von Synapsen,
- Nervenzellverlust vornehmlich im Kortex, Hippocampus und der Amygdala
- Ablagerung von extrazellulären β-Amyloid ("Plaques") und
- Auftreten von neurofibrillären Veränderungen ("Neurofibrillenbündel").
Die beiden letzteren histologischen Veränderungen stehen im Mittelpunkt der Ursachenforschung und trennen die Forscher in zwei Lager, die Anhänger der "tau-Hypothese" und der "Amyloid-Hypothese". Diese beiden Hypothesen sollen in Kürze dargestellt werden.
Die tau-Hypothese
In den neurofibrillären Veränderungen ("Neurofibrillenbündel"; "tangles") finden sich intrazelluläre "Ausfällungen", die in einer für die Alzheimer-Krankheit typischen Art und Weise im Gehirn verteilt sind. Ultrastrukturell bestehen die neurofibrillären Veränderungen aus paarig angeordneten helikalen Filamenten [4]. Beide Filamenttypen entstehen durch übermäßig oder abnorm phosphoryliertes tau-Protein. Tau gehört zu der Gruppe der Mikrotubulus-assoziierten Proteinen, deren primäre Funktion in der Stabilisierung der Mikrostruktur der Stützproteine ("Mikrotubuli") in den Nervenzellausläufern liegt. Durch die unphysiologische Phosphorylierung löst sich das an die Mikrotubuli angelagerte tau-Protein von den Mikrotubuli und es bilden sich die paarigen Filamente, die in den neurofibrillären Veränderungen als "Ausfällungen" nachgewiesen werden können. Als unmittelbare Folge gehen dann die Nervenzellen zugrunde. Wie in verschiedenen Untersuchungen nachgewiesen werden konnte, besteht ein enger Zusammenhang zwischen dem Auftreten und der Lokalisation der neurofibrillären Veränderungen und der Ausprägung der Alzheimer-Krankheit.
Kürzlich konnte nachgewiesen werden, dass einige seltene Demenzformen durch Mutationen im tau-Gen bedingt sind, wie die FTDP-17 (fronto-temporale Demenz und Parkinsonismus assoziiert mit Chromosom 17). Alle bisher bekannten Gendefekte liegen in der Domäne des Proteins, die an die Mikrotubuli bindet, was die Vermutung bestärkt, dass hier der Schlüssel für die veränderte Funktion des tau-Proteins liegt. Weitere Erkrankungen sind mit Veränderungen im tau-Protein in Zusammenhang gebracht worden und werden unter den Begriff der "Tauopathien" (z. B. progressive supranukleäre Blickparese) subsummiert.
Die Amyloid-Hypothese
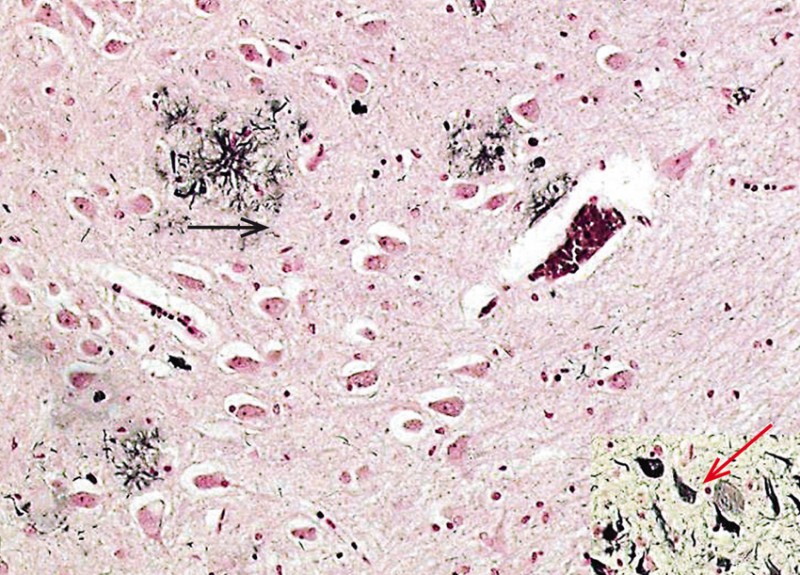
Bei der Amyloid-Hypothese steht ein kurzes Peptid, das sogenannte ß-Amyloid, das in Form von Plaques im Gehirn abgelagert wird, im Mittelpunkt des Interesses (s. Abb. 2).
Abb. 2: Typische histopathologische Veränderungen mit Amyloid-Plaque (schwarzer Pfeil) und Neurofibrillenveränderungen (roter Pfeil).
Foto: Prof. Dr. R. Dodel, Marburg
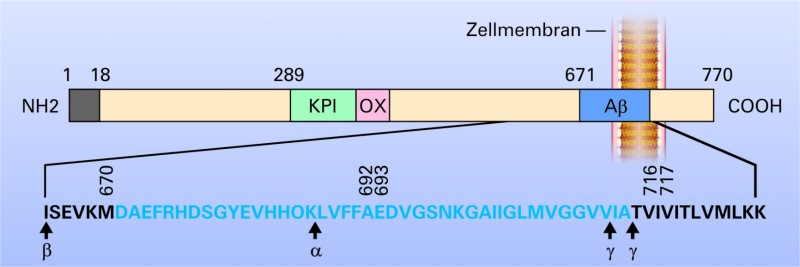
Amyloid-Plaques sind sphärische, multizelluläre Verdichtungsherde, die häufig einen zentralen Amyloid-Kern aufweisen. Sie bestehen aus geschwollenen prä- und postsynaptischen Endigungen von untergegangenen Nervenfaserfortsätzen, abnormen Synapsen, Mikroglia, Astrozyten, Makrophagen, Proteinen [5]. Die Hauptkomponente der Plaque ist ein 39-43 Aminosäuren langes Peptid (ß-Amyloid (Aß))[6]. Es entsteht durch enzymatische Spaltung des Amyloid-Präkursor-Proteins (APP), das kodiert wird durch ein Gen auf dem langen Arm von Chromosom 21. Drei Enzyme, die zur Spaltung des APP führen, die sogenannten Sekretasen (Alpha-, Beta- und Gamma-Sekretase) sind bisher nachgewiesen worden, die N-terminal, C-terminal oder mitterminal ihre Aktivität entfalten (siehe Abb. 3).
Abb. 3:
Die Entstehung von Aβ aus dem Amyloid-Präkursor-Protein (APP).
Dargestellt ist die längste der bekannten APP-Formen (APP770). Ein 17-Aminosäuren langes Signalpeptid ist am NH2
-terminalen Ende (links im Bild) vorhanden. Die Aβ-Region ist blau angefärbt. Die Wild-Typ Sequenz des Aβ ist als Einzelbuchstaben-Code angegeben und blau unterlegt. Die Zahlen über der Sequenz geben einige der bisher bekannten APP-Mutationsstellen an, die zur Alzheimer-Krankheit oder zur Erkrankung der "hereditary cerebral hemorrhage" führen. Die Schnittstellen der verschiedenen Sekretasen (α-, β-, γ-Sekretasen) sind eingezeichnet. Mittels β-Sekretase entsteht durch enzymatische Spaltung aus dem Vorläuferprotein, das sAPP-β und ein C99-Fragment, welches durch die γ-Sekretase zu Aβ gespalten werden kann. Durch Aktivierung der α-Sekretase entsteht lösliches sAPP und ein C83-Bruchstück, welches durch γ-Sekretase nochmals zu einem kleineren Protein (p3) gespalten werden kann. Aβ wird dann nicht gebildet.
ZM: Zellmembran; KPI: Kunitz-ähnlicher Proteinase-Inhibitor; OX: OX-2 Antigen-Domäne
Im Gehirn des Menschen entsteht zu ca. 85% Aß1-40 und zu ca. 5-10% Aß1-42. Dieses β-Amyloid lagert sich zusammen und bildet höhermolekulare Einheiten, sogenannte Oligomere und Fibrillen unterschiedlicher Struktur [7]. Im Gegensatz zu früheren Anschauungen, die die Plaques als Hauptursache für die Entwicklung der Alzheimer-Krankheit angesehen haben, werden diese in verschiedenen Konfigurationen und Größen vorkommenden Oligomerstrukturen derzeit als hochtoxische Produkte des Aß angesehen, die am Zelltod durch verschiedene Mechanismen beteiligt sein sollen [25]. Welche Mechanismen aber letztendlich durch die Aß-Ablagerung angestoßen werden, die dann zum Nervenzelltod führen, ist derzeit noch unklar.
Die "Amyloid-Kaskaden-Hypothese" wird durch eine Vielzahl von Befunden gestützt. So führen beispielsweise Mutationen im APP-Gen zu familiären Formen der Alzheimer-Krankheit. Deshalb zielen derzeit die meisten therapeutischen Forschungsansätze auf eine Beeinflussung der Amyloid-Kaskade hin. Zum gegenwärtigen Zeitpunkt ist noch unklar, ob die beschriebenen Veränderungen im tau-Protein oder die Ablagerung des ß-Amyloids die primären Veränderungen darstellen, die schließlich die Störungen bedingen, die zu den klinischen Veränderungen führen, die wir als Symptome der Alzheimer-Krankheit kennen.
Immunisierungsansätze zur Behandlung der Alzheimer-Krankheit
Kürzlich konnte in einem transgenen APP-Mausmodell, in dem eine ausgeprägte Plaquebildung auftritt, gezeigt werden, dass eine aktive Immunisierung mit ß-Amyloid (Aß1-42) zu einer signifikanten Abnahme der Amyloid-Plaque-Bildung und Astrogliose bei jungen Mäusen führt und bei älteren Mäusen die Plaque-Menge verringert [8]. Der Amyloid-Antikörper-Komplex wird dann von Mikrogliazellen aufgenommen und verstoffwechselt. Auch die direkte Gabe von Antikörpern (passive Immunisierung), die gegen ß-Amyloid gerichtet waren, führte zu einer deutlichen Abnahme der Amyloid-Bildung in den Gehirnen von transgenen Tieren [9]. Neben der Abnahme der Plaque-Bildung konnte nach aktiver, aber auch nach passiver Immunisierung ebenfalls eine deutliche Besserung in verschiedenen Verhaltenstests bei den transgenen Tieren festgestellt werden [10]. Dies war auch nachweisbar, wenn Tiere erst nach Ausbildung der Amyloid-Plaques immunisiert wurden. Wie die Abnahme der Plaque-Ablagerung in Anwesenheit der zirkulierenden Antikörper stattfindet, wird derzeit noch rege erforscht [11]. Unter anderen konkurrieren zwei Hypothesen:
Die Antikörper treten in geringer Konzentration in das ZNS über und führen dort zu einer Bindung des ß-Amyloids an die Antikörper und werden dann über Glia-Zellen aufgenommen und verstoffwechselt.
Es besteht ein Equilibrium zwischen Aß im Liquor und im Blut. Durch Gabe von Antikörpern, die Aß erkennen, wird das Equilibrium in Richtung Blut verschoben und mittels eines Transportmechanismus wird Aß vom ZNS in das Blut gebracht ("peripheral sink hypothesis"). Im Blut bindet es dann an die zirkulierenden Antikörper und wird dann dort verstoffwechselt [12]. Welcher Mechanismus im Patienten dann endgültig die wichtigere Rolle spielt, ist derzeit nicht klar.
Diese sehr ermutigenden tierexperimentellen Ergebnisse wurden rasch in eine klinische Studie umgesetzt. Ende 2001 wurde erstmalig mit einer aktiven Immunisierungsstudie bei Alzheimer-Patienten mit AN1792 (Aß1-42) begonnen, nachdem an einem Kontrollkollektiv mit jungen Patienten zuvor bereits eine Verträglichkeitsstudie durchgeführt worden war. Im Frühjahr 2002 traten in dieser Studie aber bei einer Reihe von Patienten schwere Nebenwirkungen ("aseptische Meningoenzephalitis") auf, so dass die Studie vorzeitig beendet werden musste [13]. Derzeit sind die hierzu publizierten Daten wenig aussagekräftig, ob die Immunisierung zu einer Besserung der kognitiven Leistung bei den Patienten geführt hat [14-16]. Inzwischen sind Patienten, die an der Studie teilgenommen haben und wegen nicht mit der Studie assoziierten Gründen verstorben sind, histopathologisch untersucht worden [17-19]. In den Gehirnen von Patienten konnte eine deutliche Abnahme der Plaquebildung nachgewiesen werden. In der Gesamtauswertung der vorzeitig abgebrochenen Studie konnte ein Trend hin zu einer verbesserten kognitiven Leistungsfähigkeit nachgewiesen werden [15, 16].
Trotz dieser ernüchternden Ergebnisse der aktiven Immunisierungsstudie mit AN1792 ist diese Forschungsrichtung "nicht am Ende" [20], vielmehr haben sich nunmehr eine Vielzahl von Ansätzen, die auf unterschiedlichen Immunisierungsansätzen basieren, entwickelt, die sich zum Teil schon in der klinischen Studienphase befinden (Tab. 2).
Tab. 2: Mögliche therapeutische Ansätze zur Immunisierung bei Morbus Alzheimer | |
Passive Immunisierung |
Aktive Immunisierung |
|
Humanisierte monoklonale Antikörper
"Humane" Antikörper
Teile von Aβ-erkennenden Antikörpern
Nanobodies
|
Adaptierte Antigene
Neue Adjuvanzien
Phagen (EFRH)
|
|
Epitope
Oligomere/Fibrillen
periphere/zentrale
Verstoffwechslung | |
Die überwiegende Mehrheit der Ansätze befasst sich mit aktiven Immunisierungsansätzen, d. h. es werden Antigene gespritzt, die im Zielorganismus zu einer Expression von gegen das Antigen gerichteten Antikörpern führt. Hier werden überwiegend Ansätze gewählt, die Aß oder Abschnitte des Aß oder dem Aß ähnelnde Konformationen als Antigen verwenden [11].
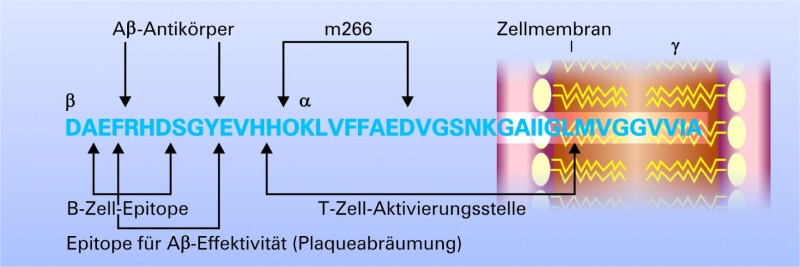
Abb. 4:
Epitope, die auf der linearen Sequenz des Aβ
bisher identifiziert wurden.
Im N-terminalen Anteil befinden sich die Sequenzen für Antikörper, die einen Effekt auf die Plaque-Deposition haben und deren Abräumung induzieren. In dieser Region liegt auch das B-Zell-aktivierende Epitop, das zu einer Induktion der B-Zellen führt. In der mitterminalen Region findet sich die Region, die zu einer Th1-vermittelten T-Zellaktivierung führen kann, die gemeinsam mit dem Immunstimulans QS-21 zu der ungewünschten Autoimmunreaktion in der klinischen Studie AN-1792 geführt haben soll [32].
M266: Epitop des Antikörpers m266; α β γ: Schnittstellen der Sekretasen.
Entscheidend für die Wahl des Antigens sind die Epitope auf dem Aß, gegen die die Antikörper gerichtet sein sollen (Abb. 4). Hier konnte gezeigt werden, dass Antikörper nach aktiver Immunisierung mit fibrilliertem ß-Amyloid ausschließlich gegen N-terminale Epitope (Aminosäuren 4-10 des Aß) gerichtet waren [21]. Diese spezifischen im N-terminalen Anteil gelegenen Antikörper waren in der Lage, Plaques abzubauen, wohingegen Antikörper, welche gegen mitterminale oder C-terminale Epitope gerichtet waren, keinen Effekt auf die Plaque-Deposition hatten. Für den gegen eine mitterminale Sequenz (AA17-24) gerichteten Antikörper m266, der derzeit von der Firma Eli Lilly entwickelt wird, konnte zwar nachgewiesen werden, dass er keine Plaques abräumen konnte, aber dennoch im Tiermodell eine Besserung der Kognition herbeiführte [22, 23]. Die Autoren führten dies auf eine Verlagerung von zentralem Aß in die Peripherie zurück ("Peripheral-sink"-Hypothese). Danach werden toxische Oligomere des Aß aus dem Gehirn entfernt und in der Peripherie dann verstoffwechselt [22, 23]. Derzeit basieren die meisten Immunisierungsansätze auf Sequenzen, die gegen den N-Terminus gerichtet sind. Eine endgültige Stellungnahme zu den "besten" Epitopen, gegen die immunisiert werden sollte, ist zur Zeit nicht möglich. In der Abbildung 4 sind die derzeit identifizierten Epitop-Sequenzen dargestellt.
Ein weiterer Ansatzpunkt bei der Immunisierung ist, ob die Antikörper nicht in Plaques abgelagertes Aß, sondern bevorzugt Monomere, Oligomere, höhergradige Oligomere oder Fibrillen des Aß erkennen [24]. Wie oben dargestellt, gibt es neuere Befunde, die darauf hinweisen, dass die frühe Generierung von toxischen Oligomeren zum neuronalen Zelltod führen und die Amyloid-Plaques einen Endzustand darstellen, mit dem das Gehirns versucht, diese zu "enttoxifizieren"[25]. Aus dieser Perspektive wäre es sicherlich wünschenswert, dass diese höhergradigen Aggregationsprodukte durch entsprechende Antikörper erkannt und verstoffwechselt werden.
Daran schließt sich direkt die Frage an, wo das Aß verstoffwechselt werden soll. In dem Ansatz der "Peripheral-sink"-Hypothese ist ein Übertritt in das Gehirn nicht unbedingt Voraussetzung, sondern die Verstoffwechslung kann in der Peripherie stattfinden. Bei dem zweiten Ansatz treten die Antikörper in sehr geringer Konzentration über die Blut-Hirn-Schranke, erkennen dort das Aβ und werden dann über aktivierte Gliazellen aufgenommen und verstoffwechselt.
Weitere Punkte, die für aktive Immunisierungsansätze neben den bereits erwähnten bedacht werden müssen, betreffen die Art der Verabreichung des Antigens sowie die Wahl des Immunstimulans. Wie sich in der Studie AN1792 gezeigt hatte, war die Wahl des Immunstimulans gemeinsam mit der Verabreichung von Aß verantwortlich für die aufgetretenen Nebenwirkungen [26].
Ein klarer Nachteil der aktiven Immunisierung ist, dass sich die Immunantwort im Alter deutlich von der Immunantwort im Kindes- oder Jugendalter unterscheidet. Bei der aktiven Immunisierungsstudie mit AN1792 konnte in nur ca. 20% der geimpften Personen eine adäquate Immunantwort erreicht werden [15]. Deshalb wird die aktive Immunisierung nicht bei allen Patienten, zumindest mit den derzeit verfügbaren Möglichkeiten, zu einer befriedigenden Therapie führen. Deshalb ist die Umsetzung auch passiver Immunisierungsansätze notwendig. Bei der passiven Immunisierung werden unterschiedliche Ansätze verfolgt, indem humanisierte Antikörper ("m266"), trunkierte oder Teile von Antikörpern oder Nanobodies entwickelt werden. Nanobodies sind Antikörper, die aus dem Tier (Lama bzw. Kamel) stammen und dadurch gekennzeichnet sind, dass sie wesentlich kleiner sind und obgleich sie nur aus einer schweren Kette bestehen, voll funktionsfähig sind [27]. Bisher ist dieser Ansatz noch nicht in der klinischen Phase. Ein weiterer passiver Immunisierungsansatz betrifft den Einsatz von humanen Immunglobulinen. Diese enthalten natürlich vorkommende Antikörper gegen Aß, die zu einer Verstoffwechslung des Aß führen können [28, 29]. Zwei unabhängige Pilotstudien an fünf und acht Patienten zeigten einen Effekt auf die Konzentration von Aß im Liquor und Serum [29, 30]. Eine 24 Patienten umfassende Dosisfindungsstudie wurde gerade in den USA abgeschlossen [31]. Erste präliminäre Daten scheinen erfolgversprechend, so dass eine große über 320 Patienten umfassende doppelblinde, randomisierte Studie mit Gabe von Immunglobulinen diesen Jahres beginnen soll.
Entscheidung in den nächsten fünf Jahren
Gegenwärtig kann aufgrund der dargestellten Therapieansätze erwartet werden, dass in den nächsten fünf Jahren eine Entscheidung getroffen werden kann, ob die Immunisierung als effektive und kausale Therapie angesehen werden kann. Da sich die Alzheimer-Krankheit als eine Folge von komplexen pathologischen Ereignissen im Laufe von Jahren oder Jahrzehnten entwickelt, bleibt die Frage nach dem Beginn einer möglichen "protektiven" Therapie. Derzeit ist Konsens, dass diese bereits bei Personen zum Einsatz kommen sollte, die sich im sehr frühen Stadium der Erkrankung befinden bzw. als Risikopersonen einzustufen sind. Leider stehen noch keine sicheren "Marker" für die Alzheimer-Krankheit zur Verfügung, die die Identifikation solcher frühen präsymptomatischen Zustände zulassen würden. Die Zukunft der Therapieforschung bei der Alzheimer-Krankheit, will sie erfolgreich sein, muss deshalb auf drei Pfeilern stehen:
1. die Testung der bereits bestehenden Therapieansätze in vorklinischen und klinischen Studien
2. Entwicklung weiterer, neuer Therapieansätze auf der Basis der Erforschung der biologischen Grundlagen der Krankheit
3. die Erforschung von "Markern", die eine frühe Identifikation der Erkrankung ermöglichen.
Dies wird in Deutschland und weltweit zum gegenwärtigen Zeitpunkt mit intensivem Aufwand untersucht, so dass der zuvor beschriebene Zeitrahmen von etwa fünf Jahren zur Entwicklung einer adäquaten Therapie für die Alzheimer-Krankheit als sinnvoll erachtet werden kann.
Korrespondenzanschrift:
Prof. Dr. Richard Dodel, Arbeitsgruppe für Neurologische Therapieforschung Neurologische Klinik, Philipps-Universität Marburg, Rudolf-Bultmannstr. 8, 35039 Marburg, E-Mail: dodel@med.uni-marburg.de
Literatur
[1] Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. Cholinesterasehemmer bei Alzheimer Demenz. Abschlussbericht A05-19A. Köln: http://www.iqwig.de/download/A0519A_Abschlussbericht_Cholinesterasehemmer_bei_Alzheimer_Demenz.pdf, 2007.
[2] Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. Memantin bei Alzheimer Demenz. Abschlussbericht A05-19C. Köln, 2007.
[3] Cummings JL, Doody R, Clark C. Disease-modifying therapies for Alzheimer disease: Challenges to early intervention. Neurology 2007; 69: 1622-1634.
[4] Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer‘s disease and related disorders. Nat Rev Neurosci 2007; 8: 663-672.
[5] Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791-1800.
[6] Selkoe DJ, Schenk D. Alzheimer‘s Disease: Molecular Understanding Predicts Amyloid-Based Therapeutics. Annu Rev Pharmacol Toxicol 2002; 4: 4.
[7] Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 2002; 30: 552-557.
[8] Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999; 400: 173-177.
[9] Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 2000; 6: 916-919.
[10] Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer‘s disease. Nature 2000; 408: 982-985.
[11] Solomon B. Beta-amyloidbased immunotherapy as a treatment of Alzheimers disease. Drugs Today (Barc) 2007; 43: 333-342.
[12] Dodel RC, Hampel H, Du Y. Immunotherapy for Alzheimer‘s disease. Lancet Neurol 2003; 2: 215-220.
[13] Monsonego A, Imitola J, Petrovic S, et al. Abeta-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of Abeta in a mouse model of Alzheimer‘s disease. Proc Natl Acad Sci U S A 2006; 103: 5048-5053.
[14] Hock C, Konietzko U, Streffer JR, et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer‘s disease. Neuron 2003; 38: 547-554.
[15] Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005; 64: 1563-1572.
[16] Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005; 64: 1553-1562.
[17] Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer‘s disease. Brain Pathol 2004; 14: 11-20.
[18] Masliah E, Hansen L, Adame A, et al. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 2005; 64: 129-131.
[19] Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 2003; 9: 448-452.
[20] Schenk D. Amyloid-beta immunotherapy for Alzheimer‘s disease: the end of the beginning. Nat Rev Neurosci 2002; 3: 824-828.
[21] McLaurin J, Cecal R, Kierstead ME, et al. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4-10 and inhibit cytotoxicity and fibrillogenesis. Nat Med 2002; 8: 1263-1269.
[22] DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer‘s disease. Proc Natl Acad Sci U S A 2001; 98: 8850-8855.
[23] Dodart JC, Bales KR, Gannon KS, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer‘s disease model. Nat Neurosci 2002; 5: 452-457.
[24] Kayed R, Head E, Sarsoza F, et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2007; 2: 18.
[25] Cheng IH, Scearce-Levie K, Legleiter J, et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 2007; 282: 23818-23828.
[26] Cribbs DH, Ghochikyan A, Vasilevko V, et al. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol 2003; 15: 505-514.
[27] Roovers RC, van Dongen GA, en Henegouwen PM. Nanobodies in therapeutic applications. Curr Opin Mol Ther 2007; 9: 327-335.
[28] Du Y, Dodel R, Hampel H, et al. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology 2001; 57: 801-805.
[29] Dodel R, Du Y, Depboylu C, Hampel H, et al. Intravenous Immunoglobulins containing antibodies against b-amyloid for the treatment of Alzheimer‘s disease. JNNP 2004; im Druck.
[30] Relkin N, Wilson R, Weksler M. Immunoglobulins for the treatment of Alzheimer‘s disease. Alzheimer Dis Assoc Disord 2006; 12.
[31] Baxter. Clinical trials of IVIG in the treatment of Alzheimer. http://wwwbaxtercom/about_baxter/news_room/news_releases/2007/08-28-07-ivightml 2007.
[32] Monsonego A, Maron R, Zota V, Selkoe DJ, Weiner HL. Immune hyporesponsiveness to amyloid beta-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer‘s disease. Proc Natl Acad Sci U S A 2001; 98: 10273-10278.



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.