














- DAZ.online
- DAZ / AZ
- DAZ 33/2011
- Der EHEC, der ein EAHEC ...
Infektiologie
Der EHEC, der ein EAHEC war
Grafik: Ilse Zündorf
Als Quelle für den Bakterienstamm EHEC O104:H4, der diese Epidemie verursachte, wurden bestimmte Chargen von aus Ägypten stammenden Bockshornkleesamen identifiziert. Der Bakterienstamm selbst gab den Medizinern und Mikrobiologen hinsichtlich seiner Infektiosität und Pathogenität Rätsel auf. Grund genug, sich nochmals genauer das Genom des Infektionsstammes anzusehen.
EHEC, EAEC oder EAHEC?
Nachdem der aus Patienten isolierte Bakterienstamm blutigen Durchfall und zum Teil auch das hämolytisch-urämische Syndrom (HUS) verursachte, wurde er sehr schnell als "enterohämorrhagischer E.-coli-Stamm" (EHEC) klassifiziert. Allerdings stellte sich bei PCR-Analysen des Genoms heraus, dass ein für enterohämorrhagische Bakterienstämme typischer Pathogenitäts-Genort fehlte. Dieser Genombereich wird auch als Locus of Enterocyte Effacement (LEE) bezeichnet und codiert üblicherweise für verschiedene Proteine, die zum einen die Adhäsion der Bakterien an die Zellen der Darmwand ermöglichen, aber auch für eine Schädigung dieser Enterozyten verantwortlich sind. Des Weiteren war erstaunlich, dass EHEC O104:H4 eine Hämolyse verursachte, obwohl die Bakterien kein Hämolysin produzieren. Stattdessen fanden sich bei dem Ausbruchsstamm diverse Gene, die typischerweise in so genannten "enteroaggregativen E.-coli-Stämmen" (EAEC) zu finden sind und beispielsweise für aggregative Adhärens-Fimbrien (AAF) codieren. Diese Fimbrien verankern einerseits die Bakterien in der Darmwand. Andererseits lösen sie auch eine Entzündung aus. Mittlerweile weiß man, dass der E.-coli-Stamm, der für die Epidemie verantwortlich war, von beiden etwas mitbekommen hatte und dadurch in seiner Gesamtheit recht infektiös und außergewöhnlich pathogen war. Als logische Schlussfolgerung kann man diese Bakterien jetzt am besten als EAHEC bezeichnen [2]. Doch, wie kam man darauf?
Vergleichende Genomanalyse
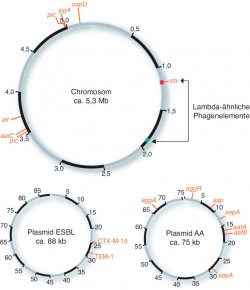
Nun kennt man schon lange sowohl enterohämorrhagische als auch enteroaggregative E.-coli-Stämme unterschiedlicher Serotypen und hat entsprechende Isolate auch in den maßgeblichen Sammlungen hinterlegt. Was lag also näher, als ein Set an unterschiedlichen Stämmen Durchfall-erregender E.-coli-Bakterien auszuwählen, deren Genom zu sequenzieren und dann mit dem Genom eines Isolats des aktuellen Ausbruchs zu vergleichen? In einer kürzlich im New England Journal of Medicine veröffentlichten Studie wurden insgesamt 13 Bakterienstämme analysiert und verglichen, die entweder enteroaggregativ, aber von einem anderen Serotyp, beispielsweise O44:H18 oder O92:H33, oder aber vom gleichen Serotyp O104:H4, jedoch nicht HUS-auslösend sind [2]. Neue Sequenziertechniken ermöglichten, dass mit drei parallel laufenden Geräten pro Isolat nur fünf Stunden Zeit benötigt wurden, um das jeweilige Genom komplett zu analysieren – ein unglaublicher Fortschritt, wenn man bedenkt, dass das Chromosom von E. coli immerhin auch ca. 5,2 x 106 bp umfasst und zusätzlich noch zwei relativ große Plasmide von ca. 75 und 90 x 103 bp im neuen EAHEC-Stamm enthalten sind (Abb. 1).
Die Idee war nun folgende: Überall da, wo sich die Genome der nicht so infektiösen, nicht so pathogenen Stämme von den Isolaten des aktuellen Ausbruchs unterscheiden, müssen genetische Informationen liegen, die für die speziellen Virulenz- bzw. Pathogenitätsfaktoren des Epidemie-Stammes verantwortlich sind.
Die Ergebnisse [2]
Shigatoxin-2-Produktion. E.-coli-Stämme des Serotyps O104:H4 sind eigentlich schon lange bekannt. Was allerdings bei diesem Ausbruch ungewöhnlich war, war die Produktion des Shigatoxin 2. Dieses Exotoxin wird eigentlich von Lambda-ähnlichen Bakteriophagen codiert und wurde erstmals im Bakterium Shigella dysenteriae entdeckt. Diese Bakteriophagen sind sehr mobile genetische Elemente und sorgen einerseits für die Ausbreitung der genetischen Information des Shigatoxins, andererseits aber auch für eine genetische Diversifikation der befallenen Bakterien. Man nimmt mittlerweile an, dass die Aufnahme des Bakteriophagen durch den O104:H4-Stamm erst kurz vor dem Epidemie-Ausbruch stattgefunden hat.
Integriert die DNA des Bakteriophagen in das Bakterien-Genom, bezeichnet man den Phagen auch als Prophage, der – ähnlich wie man es auch von HIV kennt – nach DNA-Replikation auf die Nachkommenzellen weitervererbt wird, ohne sie zunächst zu schädigen. Unter bestimmten Bedingungen kann dieser Prophage allerdings reaktiviert werden und die Zelle zur Produktion neuer Bakteriophagen stimulieren, was dann letztlich auch zur Lyse der Wirtszelle führt.
Im Genom des Isolats fand man bei der Sequenzierung zwei Integrationsorte eines Lambda-ähnlichen Prophagen. Ein Integrationsort war auch in den anderen Vergleichsstämmen vorhanden und beherbergt ein vergleichsweise harmloses Phagengenom. Der andere Integrationsort war jedoch nur in den Epidemie-Isolaten nachweisbar. Hier hatte sich ein Prophage eingenistet, der nicht nur das Gen für das Shigatoxin 2 bereitstellte. Zusätzlich fand man in den benachbarten Sequenzen Bereiche, die für die Kontrolle der Toxinproduktion und -freisetzung verantwortlich sind, so dass bei der Reaktivierung des Prophagen und Einleitung des lytischen Zyklus auch gleichzeitig noch vermehrt Shigatoxin 2 produziert wird.
Man weiß schon längere Zeit, dass beispielsweise das bakterielle "SOS"-System, das bei einer Schädigung der bakteriellen DNA oder aber bei Inhibition der DNA-Replikation induziert wird, ein starker Aktivator für den Prophagen ist. Ein sehr cleveres System, schließlich würde der Untergang der Bakterienzelle auch den Prophagen mit zerstören! Mit der Kopplung des bakteriellen Reparatursystems an die Aktivierung des Prophagen können hingegen beim ersten Alarmsignal noch schnell neue Bakteriophagen gebildet und freigesetzt werden – der Bakteriophage bleibt am "Leben". Nun weiß man auch, dass z. B. Mitomycin C, Trimethoprim-Sulfamethoxazol, Ciprofloxacin und andere Antibiotika sehr effizient das bakterielle "SOS"-System aktivieren und darüber dann bei diesem Prophagen letztlich die Shigatoxin-2-Produktion steigern können. Dies zeigte sich klinisch daran, dass Patienten, die mit einem Shigatoxin-produzierenden E.-coli-Stamm infiziert waren und mit Mitomycin C behandelt wurden, bevorzugt HUS entwickelten [3, 4]. Auch bei der O104:H4-Epidemie wurde die Beobachtung gemacht bzw. wurde befürchtet, dass der Krankheitsverlauf durch Antibiotika-Gabe negativ beeinflusst werden würde, so dass die Gesellschaft für Infektiologie e.V. am 1. Juni 2011 eine "Therapieempfehlung zu EHEC und Antibiotikabehandlung" veröffentlichte, in der "der Einsatz von Fluorchinolonen, Cotrimoxazol, Aminoglykosiden und Fosfomycin bei Patienten mit EHEC nicht empfohlen" wird. Bei gegebener Indikation sei allerdings eine Behandlung mit einem Carbapenem oder neueren Makroliden, Rifampicin oder Rifaximin vertretbar [5].
Produktion der SPATE-Virulenzfaktoren. SPATE ist die Abkürzung für "Serinprotease-Autotransporter" und steht für zwei Klassen von sezernierten Proteasen, die über einen Autotransportmechanismus durch die äußere Membran des gram-negativen Bakteriums ausgeschleust werden. Während die Klasse-I-SPATEs zytotoxisch auf Epithelzellen wirken, sind die Klasse-II-SPATEs durch ihre schleimauflösende Wirkung charakterisiert. Im O104:H4-Epidemiestamm wurde eine Kombination von drei SPATE-Genen (pic und sigA auf dem Genom, sepA auf dem Plasmid AA) nachgewiesen, die sowohl zur Klasse I als auch zur Klasse II zu zählen ist (Tab. 1).
Tab. 1: Vermutliche Virulenzfaktoren im EAHEC O104:H4 (mod. nach [2]) | ||
Gen |
Lokalisation |
Vermutliche Virulenzfunktion |
Toxin-Gene | ||
stx2a |
Chromosom |
Shigatoxin (Stx); A-B-Typ Toxin, das die Proteinbiosynthese in eukaryontischen Zellen inhibiert |
sigA |
Chromosom |
IgA-Protease-ähnliches Homolog aus S. flexneri
2a; gehört zu SPATE-Klasse-I-Genen |
pic |
Chromosom |
Mucinase; vermittelt Hämagglutination von Erythrozyten; gehört zu SPATE-Klasse-II-Genen |
sepA |
pAA-Plasmid |
Induziert Mucosa-Atrophie und Gewebeentzündung; gehört zu SPATE-Klasse-II-Genen |
aggR und AggR-regulierte Faktoren | ||
aggR |
pAA-Plasmid |
Transkriptionsfaktor für eine Reihe von Genen, die für Virulenzfaktoren codieren |
aatPABCD |
pAA-Plasmid |
ABC-Protein für den Transport von Dispersin über die äußere Membran gram-negativer Bakterien |
aap |
pAA-Plasmid |
10 kDa Disperin zur Verteilung der Bakterien über die intestinale Mucosa |
aggABCD |
pAA-Plasmid |
Aggregative Adhärenz-Fimbrien (AAF) zur Verankerung der Bakterien an die intestinale Mucosa; vermittelt Hämagglutination von Erythrozyten |
aaiA-P |
Chromosom |
Bakterielles Sekretionssystem mit unbekannter Funktion, im chromosomalen pheU-
Pathogenitätslocus lokalisiert |
Andere Gene | ||
air |
Chromosom |
Möglicherweise: Aggregation und Adhärenz |
capU |
pAA-Plasmid |
Hexosyltransferase-Homolog |
ETT2-Gene |
Chromosom |
Möglicherweise: Sekretionssystem |
Ungewöhnlich an dieser Kombination ist, dass sie bisher eigentlich nur selten in enteroaggregativen E.-coli-Stämmen beobachtet wurde. Außerdem kommen in EAEC-Stämmen meist nur zwei SPATE-Gene vor, so dass die im Epidemie-Stamm vorliegende Anzahl und Kombination die extreme Virulenz der O104:H4-Bakterien erklären könnte [2, 6].
Plasmid AA und enteroaggregative Virulenzfaktoren. Der Grund, weshalb EHEC O104:H4 doch eher zu einem EAEC wurde, liegt an dem ca. 75 kb großen Plasmid, das im Epidemie-Stamm gefunden wurde. Dieses Plasmid ähnelt sehr dem Plasmid pAA, das typischerweise in enteroaggregativen Bakterien vorkommt und für eine Reihe von Virulenzfaktoren codiert. Eine dominante Rolle spielt dabei das Genprodukt AggR, das als Transkriptionsfaktor die Expression anderer Gene von wichtigen Virulenzfaktoren wie aggABCD, aap, aatPABCD und aaiA-P steuert (Tab. 1). Die Fimbrien, also die Strukturen auf der Bakterienoberfläche, die für eine Verankerung der Bakterien an der intestinalen Mucosa sorgen, eine Hämagglutination der Erythrozyten und Entzündungsreaktionen auslösen, werden vom Operon aggABCD codiert. Durch diese Fimbrien sieht ein Darmbefall mit enteroaggregativen Bakterien aus wie mit gestapelten Ziegelsteinen belegt (stacked-brick). Damit dann aber doch die Bakterien ein wenig über die intestinale Mucosa verteilt werden, sezernieren EAECs das 10 kDa große und vom aap-Gen codierte Protein Dispersin, das wiederum über einen speziellen, vom aatPABCD – Operon codierten Transportmechanismus über die äußere Membran der Bakterienzelle ausgeschleust wird (Tab. 1).
ESBL-Plasmid. Neben dem pAA-Plasmid wurden noch ein mit 1,5 kb recht kleines und ein ca. 88 kb großes Plasmid gefunden, die nicht für Virulenzfaktoren codieren. Das kleine Plasmid scheint bedeutungslos zu sein, das große Plasmid wurde als pESBL bezeichnet. Der Vergleich zwischen den untersuchten Bakterienstämmen zeigte, dass die meisten anderen O104:H4-Stämme pESBL nicht enthielten. Entweder ist also pESBL instabil und geht relativ schnell verloren, oder aber einige O104:H4-Stämme haben das Plasmid erst relativ frisch aufgenommen.
ESBL ist die Abkürzung für "Extended-Spectrum Beta-Lactamases" und steht für Lactamasen, die z. B. Cephalosporine der dritten Generation spalten können und von den Resistenz-Genen TEM-1 und CTX-M-15 codiert werden. pESBL ähnelt sehr stark dem Plasmid pEC_Bactec, das in verschiedenen klinischen E.-coli-Isolaten gefunden wurde und für eine Ausbreitung der Antibiotikaresistenz zwischen unterschiedlichen Krankenhauskeimen mitverantwortlich ist.
Pathogenesemechanismus
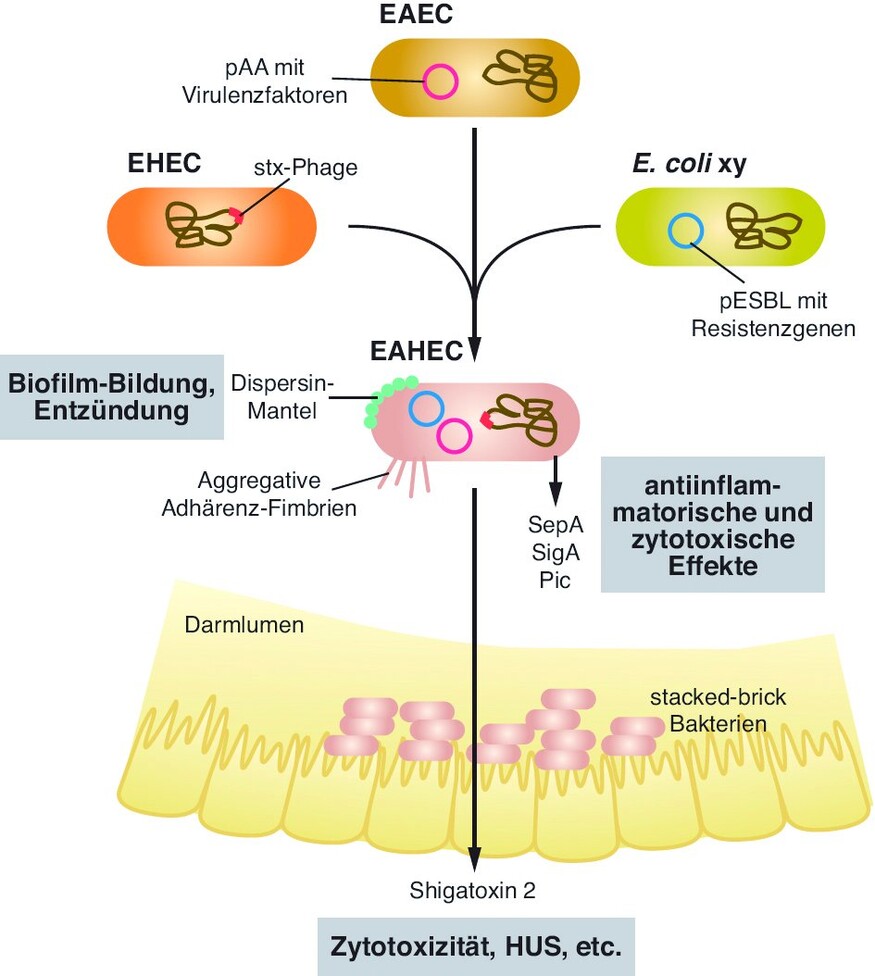
Das hinsichtlich Infektiosität und Pathogenität ungewöhnliche EAHEC O104:H4 hat mittlerweile verschiedene Forschergruppen ermutigt, die speziellen Virulenzfaktoren des Bakterienstammes aufzuklären (beispielsweise [2, 7– 10]). Inzwischen sind mehrere Isolate aus Patienten sequenziert und analysiert worden, und man sieht, dass sich doch ein leicht heterogenes Bild hinsichtlich einzelner Virulenz- und Resistenzfaktoren bei den verschiedenen Isolaten ergibt. Die hier vorgestellten, wichtigsten Toxine und Virulenzfaktoren lassen jedoch auf folgende erste Infektionsschritte schließen [7]:
Über die aggregativen Adhärenz-Fimbrien (AAF) heften sich die Bakterien an die intestinale Mucosa.
Die AAF vermitteln weiterhin die autoaggregative Adhäsion, die dazu führt, dass sich die Bakterien wie gestapelte Ziegelsteine aufeinanderlagern und einen Schleim-vermittelten Biofilm auf der Enterozytenoberfläche bilden.
Die von den Bakterien freigesetzten Toxine induzieren eine Entzündungsreaktion, beeinflussen die intestinale Sekretion und führen zur Zytotoxizität bei den Enterozyten der Darmwand.
Wo kommt dieser Bakterienstamm her?
Wie so oft, wurde ja auch bei dieser Epidemie zum Teil wild spekuliert, ob das Bakterium Menschenwerk war, ob Gentechniker ihre Finger im Spiel hatten und diesen Bakterienstamm künstlich hergestellt haben. Betrachtet man aber die genetischen Elemente, über die die verschiedenen Virulenz- und Resistenzfaktoren codiert werden, sind es überwiegend Transposons, Bakteriophagen, Plasmide oder so genannte Pathogenitätsinseln, die sich hervorragend über den so genannten horizontalen Gentransfer – also der Weitergabe zwischen nebeneinander lebenden Bakterien beispielsweise über Konjugation – verbreiten lassen.
Die Rekonstruktion des Ablaufs sieht so aus, dass in diesem Fall der Ausgangsstamm wahrscheinlich ein EAEC war, der von einem Shigatoxin-Bakteriophagen eines EHEC infiziert wurde und schließlich auch noch das Resistenzplasmid von einem Krankenhauskeim übernommen hat (Abb. 2).
Grafik: Ilse Zündorf
Resümee
Diese Epidemie hat erstaunlicherweise nur drei Monate angedauert: von Anfang Mai bis – offiziell – Ende Juli. Die gefühlte Dauer während der Epidemie war allerdings wesentlich länger, haben sich doch die Meldungen über die verschiedenen, möglicherweise kontaminierten Lebensmittel überschlagen und zu einer völligen Verunsicherung der Bevölkerung geführt. Mittlerweile ist sie jedoch schon wieder völlig aus den Köpfen der Leute verschwunden.
Was können wir aus diesem Fall lernen?
Zum einen handelte es sich um eine Kontamination mit E.-coli-Keimen, die bei ausreichender Hygiene wohl sicher vermeidbar gewesen wäre.
Durch modernste molekularbiologische und mikrobiologische Verfahren ist es gelungen, die Entstehung dieses Erregerstammes nachzuzeichnen und deutlich zu machen, wie gefährlich und rücksichtslos die Natur sein kann, wenn man mit ihr leichtfertig umgeht. Und wir sollten gewarnt sein, denn jederzeit kann sich etwas Ähnliches wiederholen.
Nachdem man O104:H4 eindeutig identifizieren konnte und vermehrt nach ihm suchte, fand man diesen Bakterienstamm an vielen Stellen, beispielsweise auch in einem Bach in der Nähe von Frankfurt/Main. Waren die Bakterien schon die ganze Zeit da und hat man nur nicht danach gesucht, oder sind sie im Zusammenhang mit der Epidemie erst dort aufgetreten? Man sollte also eigentlich immer daran denken, dass Bakterien überall vorkommen, weshalb es so wichtig ist, Obst und Gemüse vor dem Verzehr gründlich zu waschen.
Wir haben ein weiteres Mal gelernt, dass es extrem schwierig ist, Epidemien "zu managen". Einerseits ist es das Recht der Bevölkerung, schnell und umfassend informiert zu werden. Andererseits steigt dadurch die Gefahr, dass die Information zu einem überschießenden Verhalten führt. Information wird oft ausgenutzt als Quelle für Sensation, und Sensation ist eine ideale Basis für Panik und Hysterie. Dass eine nahezu auf Norddeutschland begrenzte, lokale Epidemie dazu führte, dass auch die Bauern in München auf ihren Gurken und Tomaten sitzen blieben, kann im Nachhinein nur Verwunderung und Erstaunen auslösen. Die Zeche zahlen nicht diejenigen, die mit der Sensation gute Gewinne gemacht haben, sondern wir alle über Solidarsysteme, in die wir mit unseren Steuermitteln ständig einzahlen.
Ob all dies tatsächlich eine Lehre ist, bleibt skeptisch abzuwarten.
Literatur
[1] http://www.rki.de
[2] Rasko, D.A., et al.: Origins of the E. coli Strain Causing an Outbreak of Hemolytic-Uremic Syndrome in Germany. N Engl J Med. 2011 Jul 27. [Epub ahead of print]
[3] Zhang, X., et al.: Quinolone Antibiotics Induce Shiga Toxin– Encoding Bacteriophages, Toxin Production, and Death in Mice. J Infect Dis 181 (2000), 664-670
[4] Acheson, D.W.K., Donohue-Rolfe, A.: Cancer-associated hemolytic uremic syndrome: a possible role of mitomycin C in relation to Shiga-like toxins. J Clin Oncol 7 (1989), 1943
[5] http://www.rki.de Empfehlungen der Deutschen Gesellschaft für Infektiologie zu EHEC und Antibiotikabehandlung
[6] Boison, N., et al.: Short Report: High Prevalence of Serine Protease Autotransporter Cytotoxins among Strains of Enteroaggregative Escherichia coli. Am J Trop Med Hyg 80 (2009), 294– 301
[7] Brzuszkiewicz, E., et al.: Genome sequence analyses of two isolates from the recent Escherichia coli outbreak in Germany reveal the emergence of a new pathotype: Entero-Aggregative-Haemorrhagic Escherichia coli (EAHEC). Arch Microbiol. 2011 Jun 29. [Epub ahead of print]
[8] Bielaszewska, M., et al.: Characterization of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect Dis. 2011 Jun 22. [Epub ahead of print]
[9] Mellmann, A., et al.: Prospective Genomic Characterization of the German Enterohemorrhagic Escherichia coli O104:H4 Outbreak by Rapid Next Generation Sequencing Technology. PLoS One. 2011;6(7):e22751. Epub 2011 Jul 20.
[10] Rohde, H., et al.: Open-Source Genomic Analysis of Shiga-Toxin-Producing E. coli O104:H4. N Engl J Med. 2011 Jul 27. [Epub ahead of print]
Autoren
Dr. Ilse Zündorf, Prof. Dr. Theodor Dingermann, Institut für Pharmazeutische Biologie, Biozentrum, Max-von-Laue-Str. 9, 60438 Frankfurt/Main
Zum WeiterlesenEHEC – eine beherrschbare Gefahr? Von Ilse Zündorf und Theo Dingermann |



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.